Mucolipidosis II (ML II) or inclusion cell disease (I-cell disease) (OMIM 252500) is an autosomal recessive lysosomal enzyme targeting disorder. ML II is usually presents between 6 and 12 months of age with a clinical phenotype resembling Hurler syndrome and a radiological picture of dysostosis multiplex. When ML II is severe enough to be detected in the newborn period, the radiological changes have been described as similar to multiple pathological fractures of osteogenesis imperfecta [1], or rickets because of transient neonatal hyperparathyroidism [2].
The patient was the first child of the non-consanguineous Japanese parents. Short lower extremities were detected by routine fetal ultrasonography. He was born at 36 gestational weeks by natural vaginal delivery at a birth weight of 1914 g. Soon after birth, the bending deformation of his limbs was noticed and a babygram was examined. Initially, osteogenesis imperfecta was suspected from multiple fractures of long bones (Panel A). However, careful evaluation of X-ray films revealed metaphyseal fraying of femora and tibiae like rickets, and cloaked appearance of the humerus suggesting hyperparathyroidism (Panel B). Along with these findings, stippling calcifications of the calcaneum and sacrococcygeal regions were noted (Panel A). The last findings are characteristic on ML II [1,2]. The hemogram showed vacuole-like inclusions in some peripheral blood lymphocytes of the patients (Panel C). Cultured skin fibroblasts of the patient showed cytoplasmic inclusions (Panel D). Many of lysosomal enzyme activities including β-D-hexosaminidase, β-D-glucuronidase, β-D-galactosidase and α-L-fucosidase were elevated in plasma and decreased in cultured skin fibroblasts. Finally, GNPTAB gene mutation analysis revealed compound heterozygous mutation of p.Arg1189* and exon2 duplication, and the diagnosis was confirmed.
For radiological diagnosis of neonatal form of ML II, differential diagnosis of other bone diseases is occasionally required to a physician. When the doctor aware of the characteristic pictures on X-ray film then confirmed lymphocytes vacuoles in the hemogram, the way to the correct diagnosis can be opened.
The author declares no conflict of interest related to this article.
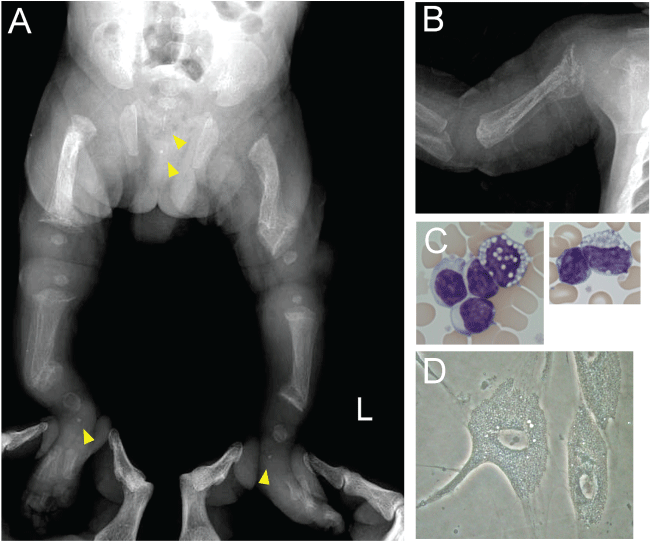
Panel A: A lower extremities radiograph showing bending femora and tibiae by pathological fractures and remodeling. Bone trabeculae are irregular, primitive and fibrillar with poor differentiation between the cortical and medullary zones. Metaphyses of these bones are frayed and expanded. Stippling calcifications are seen in bilateral tarsal and sacrococcygeal regions (yellow arrow heads).
Panel B: A right humerus radiograph showing cloaked appearance (bone within a sleeve of bone).
Panel C: Peripheral blood lymphocytes of the patients have vacuole-like inclusions (May-Giemsa staining, original magnification, ×1,000).
Panel D: Cytoplasmic inclusions (I-cell phenotype) in cultured skin fibroblasts (Phase-contrast microscopy, original magnification, ×100).