Juvenile HD (JHD) is the result of genetic anticipation that occurs due to instability of CAG expanded alleles (HTT gene) when passed to the next generation, resulting in earlier onset of clinical manifestations in successive generations.
The aim of this study was to report a Brazilian case of JHD with paternal transmission.
We evaluated a patient with JHD with chorea, cervical dystonia and aggressiveness. The Unified Huntington' Disease Rating Scale (UHDRS) was applied. The size of CAG allele was double-checked in two laboratories.
We report a patient with paternally inherited JHD caused by a large expansion (74 CAG). The patient was prescribed olanzapine 2.5 mg q.d., oxcarbazepine 300 mg bid and underwent botulinum toxin treatment, with partial improvement of aggressiveness and chorea and partial resolution of torticollis. The MRI revealed bilateral caudate and putamen atrophy. JHD is rarer than adulthood HD, with an average prevalence rate of 5%. UHDRS has limited efficacy in evaluating JHD patients independence and functional capacity, as it was originally developed for adult subjects. There is currently no specific scale for JHD patients.
CAG repeats, Juvenile Huntington's disease, UHDRS, Anticipation
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder characterized by motor, cognitive and behavioral symptoms [1,2]. The prevalence of the disease varies according to the ethnic origin: 5-10/100,000 among Caucasians from Western Europe [3-5] to 0.40/100,000 in the Asian population, for example [6]. HD is caused by a dynamic mutation due to expansion of CAG track in the HTT gene on chromosome 4. The number of CAG trinucleotide repeats in normal individuals ranges from 6 to 35. Among patients with HD, the size of CAG alleles may range from 36 to 250 repeats. The most characteristic symptom in HD is chorea, which may develop gradually over years, but patients, particularly those with early disease onset, can eventually develop other movement disorders, such as parkinsonism, dystonia, and myoclonus. The length of the CAG track is inversely proportional to the age of clinical symptoms onset [7,8]. In most cases, individuals with more than 60 CAG repeats develop the juvenile form of HD (JHD), because their symptoms begin before age 20 [9]. JHD is the result of genetic anticipation, that occurs due to the instability of CAG expanded alleles when passed to the next generation, resulting in earlier onset of clinical manifestations in successive generations [10,11]. Concerning allelic instability, there is a greater tendency to expand the CAG track when paternal alleles are transmitted to the offspring, due to the numerous rounds of DNA replication previous to spermatogonium mitosis during spermatogenesis [12]. Some authors have suggested that large expansion of CAG in HD does not occur with a frequency much greater than that predicted by normal variation when maternal origin of the expansion is being considered [13]. In Brazil the incidence of JHD is unknown. The aim of this study was to report a Brazilian case of JHD generated by an unusual CAG expansion in one paternal transmission.
This study was approved by the Research Ethics Committees registered with CAAE number 26387113.1.0000.5258. We evaluated the patient using the Unified Huntington' Disease Rating Scale (UHDRS), which is a clinical rating scale used to assess motor function, cognitive function, behavioral abnormalities and functional capacity in patients with HD. In motor and behavioral evaluation, the score is directly proportional to the severity of the disease, while when assessing functional capacity and independence, lower scores mean higher impairment. The patient herein reported was recruited at Neurology Institute of Federal University of Rio de Janeiro (UFRJ) and the genetic test was performed at the Department of Genetics and Molecular Biology of Federal University of the State of Rio de Janeiro (UNIRIO) and confirmed at the Molecular Genetics Department of Hermes Pardini Laboratory. The size of CAG allele was double-checked in two laboratories with different pair of primers because the length of the HD expanded allele was greater than usual (> 65 CAG repeats). The size of normal allele was determined in laboratory 1 (UNIRIO) and 2 (Hermes Pardini) and were coincident (18 CAG repeats). As for the expanded alleles, lab 1 detected 72 CAG repeats and lab 2 detected 74 repeats. The 74 allele was validated with samples of known sizes just in lab 2, because controls with expanded alleles larger than 63 CAG repeats were unavailable in lab 1. The pedigree was built using the free software GenoPro (2007 version). A questionnaire was used to collect the participant's personal data. The analysis of polymorphic regions of CAG repeats in the HTT gene was conducted using PCR-STR fluorescence assay. The primers and the PCR conditions used at lab 1 were according to [14] adding a Sanger sequencing validation step using different sizes of CAG alleles as a positive control. In lab 2, the PCR reaction followed by fragment analysis were performed. The PCR reaction consisted of: 10 μM forward and reverse primers, 1x STR PROMEGA buffer, 0.7X Enhancer Invitrogen, 4U Taq DNA polymer Platinum Invitrogen in 14 uL reaction. The primers were designed and commercialized by Hermes Pardini laboratory, so these sequences will not be disclosed. The amplicon was analyzed by an automated capillary electrophoresis system in ABI3730 and ABI3130xL equipment (Applied Biosystems, Foster City, CA, USA).
A 17-year-old brazilian male was evaluated in our Movement Disorders outpatient clinic with psychiatric symptoms and involuntary cervical movements, which had begun at the age of 14. Psychiatric symptoms included agitation and aggressiveness. On exam he had spasmodic torticollis, grimacing, hypotonia, and slow saccades. There was no parkinsonism, chorea, motor impersistence or oculomotor apraxia. The UHDRS score was as disclosed in the Table 1. His Mini-mental status examination score was 23 out of 30, with lost points in executive function and calculation. The patient was started on olanzapine 2.5 mg q.d., oxcarbazepine 300 mg bid and botulinum toxin for the cervical dystonia, with partial improvement. The effect resulting from the use of the drugs is shown in Table 2. In the following three years he developed severe dysartria, mild dysphagia, worsening of dystonia, paroxistic myoclonus and severe postural instability with falls requiring constant support to walk, as well as progressive cognitive decline, becoming dependent on most activities of daily living. Choreiform movements appeared five years after the onset of the disease, and affected the trunk, limbs and tongue. His father had chorea and dementia, and a genetic assay confirming the diagnosis of HD, with 50 CAG repeats. His grandfather also had similar symptoms but was not genetically tested (Figure 1). The patient' genetic test confirmed the diagnosis of HD in heterozygosity with 18/74 CAG repeats, with an uncommon paternally transmitted expansion (+24 CAG). Brain MRI showed bilateral high signal in T2 and FLAIR weighted-images, and atrophy of caudate and putamen (Figure 2).
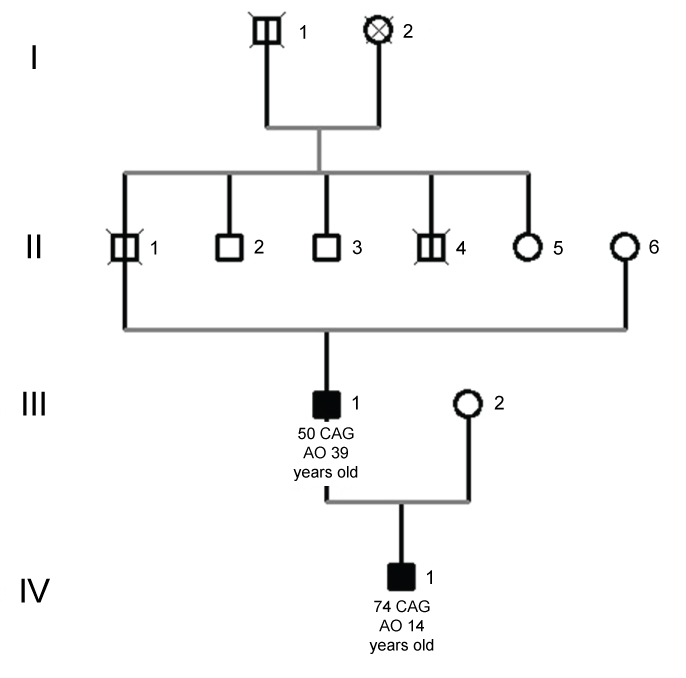 Figure 1: Juvenile patient's pedigree. Legend: AO (age of onset). Symbols in black mean subjects affected by HD and symbols marked with a vertical black line represent individuals reported with HD clinical symptoms but not genetically diagnosed. Circle: female; Square: male. View Figure 1
Figure 1: Juvenile patient's pedigree. Legend: AO (age of onset). Symbols in black mean subjects affected by HD and symbols marked with a vertical black line represent individuals reported with HD clinical symptoms but not genetically diagnosed. Circle: female; Square: male. View Figure 1
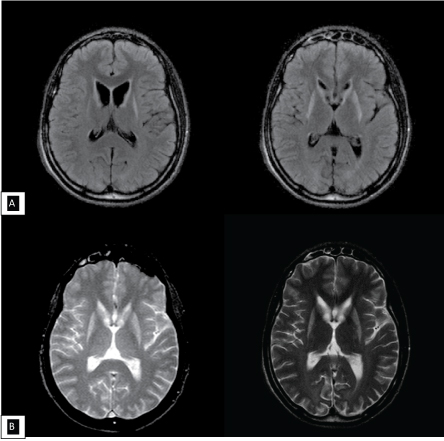 Figure 2: Axial FLAIR (A) sequence and T2 weighted-images (B) performed at 18 years old revealing bilateral caudate and putamen atrophy with hyperintensities. View Figure 2
Figure 2: Axial FLAIR (A) sequence and T2 weighted-images (B) performed at 18 years old revealing bilateral caudate and putamen atrophy with hyperintensities. View Figure 2
Table 1: UHDRS score. View Table 1
Table 2: Medication and effects. View Table 2
Patients with HD typically show the first symptoms around 40 years old. In about 2% of the cases, including ours, the onset occurs before the age of 20, which defines JHD [15]. Patients with this diagnosis usually have more than 60 CAG repeats, as opposed to 40-50 repeats in adult-onset HD [16]. JHD is rarer than adulthood HD, with an average prevalence rate of 5% of the overall HD prevalence. [17]. In 2000, Raskin, et al. [18] reported in another Brazilian study 2/84 of JHD cases. In 2012, Agostinho, et al. [14] reported in a Brazilian cohort only one case of juvenile disease (1/61). JHD is paternally inherited from in 80-90% of these cases [19]. Although other juvenile cases are not the focus of this work, it is worth mentioning that seven out of 75 (9.3%) individuals of our HD database had the first symptoms between 21 and 28 years old, and three (4%) started between the ages of 14 and 18 years. Among HD patients with disease onset before age 19, only two had more than 60 CAG repeats, this includes our patient with 74 CAG repeats and age of onset at age 14. Therefore, the frequency of JHD in our sample was 2.7% (2/75). Among all the cases (n = 10) that started HD before 29 years old, it was possible to determine the CAG alleles of affected parents in three intergenerational transmissions. Three uncommon expansion events were observed with +8, +12 and +24 CAGs (this study). Only 1/10 of these individuals were assessed by UHDRS (this juvenile case report), the others 9/10 had behavior symptoms (aggressiveness and depression), cognitive disorder, chorea, weight loss over 7 kg, dysarthria and dysphagia, as well as learning disability at school. The largest reported intergenerational expansion had +160 units, wherein a father with 54 CAG repeats had a child with 214 [20]. The first large study of HD in Brazil reported two siblings with JHD, they manifested HD with 5 and 11 years old and their expanded alleles had 88 and 69 CAG repeats respectively. Another JHD case was reported in this same study in one paternal transmission with an expansion of 41 CAG repeats (father with 47 and daughter with 88 units) in one generation, the largest paternally transmitted expansion reported in Brazil [18]. The evaluation of a JHD case must consider the behavioral, cognitive and motor abnormalities and careful interviews of relatives and caregivers by a specialist with expertise in HD [17]. Our patient had early dystonia and did not show choreiform movements until later stages. In JHD dystonia is much more frequent than in adult-onset cases. In a series by Ribaï, et al. dystonia was the most common motor manifestation among 29 JHD patients, occurring in 72% [21]. In the same study, chorea was the initial sign in 10%. Early psychiatric manifestations are common in JHD. They are reported as the first sign of disease in 65.5% of patients [21], which may result in later recognition of the disease, leading to delayed diagnosis and unnecessary testing [22,23]. In our patient, the early behavioral symptoms led to school problems and a definite diagnosis was possible only 3 years after disease onset. While brain MRI in adult-onset HD typically shows caudate atrophy, in the juvenile form putaminal atrophy is also common [24,25]. Atypical symptoms are more frequent in JHD such as bradykinesia, dystonia, cerebellar signs, and seizures [26-28]. The clinical evaluation by UHDRS has limited efficacy in testing JHD patients independence and functional capacity, as this scale was originally described for adult subjects with HD. Currently there is not a specific scale to JHD validated in Brazil. There are still many unsolved issues related to JHD onset determination, frequency, and clinically and imaging characterization. We believe this case report may contribute to the expansion of knowledge about JHD rare variant. Furthermore, there is a limited number of reports focusing on JHD from South America, except the Maracaibo lake study from Venezuela.
This work was supported by CAPES/PROAP and FAPERJ. There is no conflict of interest in this paper.