The solution for the so-called "calcium paradox" has been revealed 4 years ago, when we demonstrated the involvement of the interaction between Ca2+ and cAMP signalling pathways (Ca2+/cAMP signalling interaction) in this enigma. The "calcium paradox" emerged 4 decades ago, when numerous clinical studies have concluded that prescription of L-type Ca2+ Channel Blockers (CCBs) for hypertensive patients decreased arterial pressure, but produced stimulation of sympathetic hyperactivity. Indeed, initially these adverse effects of CCBs have been attributed to adjust reflex of arterial pressure, but this conclusion remained not completely satisfactory. The year of 2013 would change this history forever! Through an original experiment, we revealed that the "calcium paradox" phenomenon came from increased transmitter release from sympathetic neurons stimulated by CCBs due to its handling on the Ca2+/cAMP signalling interaction. It is now well-established that the signalling pathways mediated by Ca2+ and cAMP can interact, thus playing a vital role in cellular processes of mammalians. In the clinical pharmacology, it has opened novel opportunities for the development of pharmaceuticals more efficient, and safer, for treating neurodegenerative diseases. Then, the manipulation of Ca2+/cAMP signalling interaction could improve therapeutic strategies for stimulating synaptic transmission compromised by transmitter release deficit, and attenuating death of neurons. More recently, the manipulation of this interaction has been proposed by us to inhibit cancer progression, another interesting avenue for medical research.
Signalling pathways mediated by Ca2+ and cAMP, "Calcium paradox", Neurology, Cancer
Since the last decade, it has been shown that the signalling pathways mediated by Ca2+ and cAMP can interact (Ca2+/cAMP signalling interaction), thus playing a vital role in cellular processes of mammalians. In the clinical pharmacology, the manipulation of this interaction could improve therapeutic strategies for stimulating synaptic transmission compromised by transmitter release deficit, and attenuating death of neurons. More recently, the manipulation of this interaction has been proposed by us to inhibit cancer progression. All this history initiated with the concept of the "calcium paradox". It has been almost 4 years since we revealed the involvement of the Ca2+/cAMP signalling interaction in the enigma of the so-called "calcium paradox". For understanding the "calcium paradox", we should return to the past. Indeed, the concept of stimulus-secretion to elucidate neurotransmitters release has been achieved from creative experiments made by Douglas and Rubin in the 1960s [1]. By their concepts, in 1970's Baker and Knight showed that an increase in the cytosolic Ca2+ concentration ([Ca2+]c) is a fundamental requirement to start transmitter release [2]. In addition, the unquestionable result showing a correlation between neurotransmitter release and elevation in [Ca2+]c came from the interesting experiments made by the Nobel laureate Erwin Neher [3]. Thus, by reducing extracellular Ca2+ through blocking Ca2+ channels, we should have a reducing in the neurotransmitter release. Nonetheless, many reports have demonstrated that L-type Ca2+ Channel Blockers (CCBs), in concentrations below 1 μmol/L, could induce neurotransmitter release, a "paradox" [4-6]. In addition, many reports have demonstrated that cAMP enhances neurotransmitter release at several synapses in autonomic nervous system of mammalians [7]. Recently, we demonstrated that Ca2+/cAMP signalling interaction is implicated in the modulation of transmitters release from sympathetic neurons, and thus in the "calcium paradox" [8-11].
It is well established that the interaction between Ca2+ and cAMP signalling pathways is as a vital cellular process in mammalians [8-11]. This classical concept assumes that these signalling pathways virtually exist in all mammalian cells, modulated by Adenylyl Cyclases (ACs) and Phosphodiesterases (PDEs) [8-11]. In addition, Endoplasmic Reticulum (ER) Ca2+ channels have particularly been a forefront for the interaction between Ca2+ and cAMP signalling pathways field, such as Ca2+ channels modulated by Ryanodine Receptors (RyR) [8-11]. We reinforced the idea that the interaction between Ca2+ and cAMP signalling pathways plays a fundamental participation in the modulation of neurotransmitter release from neurons and neuroendocrine cells [8-11]. Then, the interaction of Ca2+ and cAMP signalling pathways could be a new therapeutic goal for pharmaceuticals.
The prescription of L-type CCBs in hypertensive patients has been reported to decrease arterial pressure, but also produces sympathetic hyperactivity [12]. Initially, these adverse effects of CCBs have been attributed to adjust reflex of arterial pressure, but this conclusion remained not completely satisfactory. The year of 2013 would change this history forever! Through a creative experiment, we revealed that the solution for this so-called "calcium paradox" phenomenon was due to the increase of transmitter release from sympathetic neurons achieved by CCBs due to its handling on the interaction between Ca2+ and cAMP signalling pathways [9]. We demonstrated that contractions of the smooth muscle (vas deferens) were completely inhibited by L-type CCBs in high concentrations (> 1 μmol/L), but puzzlingly increased in concentrations below 1 μmol/L, thus defined as sympathetic hyperactivity promoted by CCBs [4-6,9]. Our studies clearly established that the contradictory sympathetic hyperactivity is due to an augmentation of transmitter release from sympathetic neurons achieved by L-type CCBs due to its interfering on the interaction between Ca2+ and cAMP signalling pathways.
In fact, many reports have shown that elevation of cytosolic cAMP concentration ([cAMP]c) reduces neuronal death resulted from cytosolic Ca2+ overload, stimulating neuroprotective effect [13,14]. As mentioned above, the L-type CCBs increase transmitter release due to its handling on the interaction between Ca2+ and cAMP signalling pathways. This interference activates ACs, causing elevation of [cAMP]c that, in turn, induces Ca2+ release from ER that stimulates transmitter release [8-11]. In addition, this elevation of [cAMP]c produces neuroprotective effects mediated by the Ca2+ and cAMP signalling pathways [8-11]. It was proposed that this neuroprotective effect results from activation by cAMP on the cellular survival pathways mediated by PKA/CREB [8-11,13,14]. Then, the pharmacological interfering of the Ca2+/cAMP signalling interaction from the combined use of the L-type CCBs prescribed in the antihypertensive therapy, and [cAMP]c-enhancer compounds prescribed in the anti-depressive therapy like rolipram, could be a novel pharmacological goal for increasing neurotransmission in neurological and psychiatric disorders resulted from deficit of neurotransmitter release, and neuronal death [8-11]. Figure 1 illustrates how the pharmacological handling of the interaction between Ca2+ and cAMP signalling pathways could produce increase of neurotransmitter release, and attenuation of neuronal death.
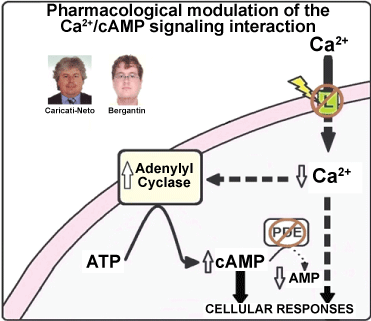 Figure 1: Transmitter release stimulation and reduction of neuronal death triggered by Ca2+ overload can be achieved due to pharmacological regulation of the interaction between Ca2+ and cAMP signalling pathways. In response to the decreasing of Ca2+ influx through L-type voltage-activated Ca2+ channels produced by CCBs, the adenylyl cyclase activity (and consequently cAMP) is increased. These CCBs-effects can be stimulated by cAMP-enhancer compounds (like PDEs inhibitors). PDEs - Phosphodiesterases, RyR - Ryanodine Receptors, IP3R - IP3 Receptors, SERCA - Sarcoendoplasmic Reticulum Ca2+-ATPase. View Figure 1
Figure 1: Transmitter release stimulation and reduction of neuronal death triggered by Ca2+ overload can be achieved due to pharmacological regulation of the interaction between Ca2+ and cAMP signalling pathways. In response to the decreasing of Ca2+ influx through L-type voltage-activated Ca2+ channels produced by CCBs, the adenylyl cyclase activity (and consequently cAMP) is increased. These CCBs-effects can be stimulated by cAMP-enhancer compounds (like PDEs inhibitors). PDEs - Phosphodiesterases, RyR - Ryanodine Receptors, IP3R - IP3 Receptors, SERCA - Sarcoendoplasmic Reticulum Ca2+-ATPase. View Figure 1
In fact, it was showed that the prescription of L-type CCBs is able to reduce motor symptoms, and reduces the continued neuronal death in animal model of Parkinson's disease, indicating that L-type CCBs are potentially workable neuroprotective pharmaceuticals [15]. Intriguingly, a 1-decade study involving thousands senile hypertensive patients demonstrated that prescription of L-type CCBs can reduce blood pressure, and incidence of dementia in hypertensive patients, indicating that these pharmaceuticals could be used to treat neurodegenerative diseases in clinics [16]. These results for the effects related to neuroprotection of CCBs have been reinvestigated in thousands elderly hypertensive patients with dysfunction of memory abilities [17]. These studies concluded that patients who have taken CCBs had their risk of cognitive dysfunction decreased, such as Alzheimer's disease [17]. These findings reinforce the concept that L-type CCBs can reduce cytosolic Ca2+ overload produced due to blocking of Ca2+ influx, and thus could be an alternative pharmacological goal to reduce, or prevent, death of neurons resulted from neurodegenerative diseases.
Based on these findings, we have anticipated that the pharmacological regulation of the Ca2+/cAMP signalling interaction by combined use of the L-type CCBs and [cAMP]c-enhancer compounds could be a novel therapeutic goal for increasing neurotransmission in neurological, and psychiatric disorders, resulted from neurotransmitter release deficit and neuronal death [8-11]. This pharmacological strategy opens a novel pathway for the drug development more efficient for the treatment of Alzheimer's and other neurodegenerative diseases [18-23,11].
In addition, it has been shown that the dysregulation of intracellular signaling pathways mediated by Ca2+ and cAMP participates in the cancer initiation, tumor formation, tumor progression, metastasis, invasion and angiogenesis. Thereby, proteins involved in these pathways, such as Ca2+ channels and cAMP-dependent Protein Kinase (PKA), represent potential drugs targets for cancer therapy [24]. With this concept in mind, some studies showed that drugs able to interfere with the intracellular Ca2+ signaling such as selective CCBs, as amlodipine, inhibit proliferative response in different cancer cells [24-27]. In addition, drugs able to increase the intracellular cAMP levels (cAMP-enhancer compounds), such as Phosphodiesterase (PDE) 4 inhibitors, have been proposed as potential adjuvant, chemotherapeutic or chemopreventive agents in some cancer types, including hepatocellular carcinoma [28]. Then, the pharmacological modulation of the intracellular signaling mediated by Ca2+ and cAMP in the cancer cells may represent a new therapeutic strategy for cancer progression.
In conclusion, pharmacological interfering of the interaction between Ca2+ and cAMP signalling pathways could be a more efficient therapeutic approach for enhancing neurotransmission resulted from neurotransmitter release deficit, and reducing neuronal death in the neurodegenerative diseases (like Alzheimer's and Parkinson's diseases). More recently, the manipulation of this interaction has been proposed by us to inhibit cancer progression, another avenue for medical research. These findings could dramatically impact in clinical pharmacology.
Caricati-Neto and Bergantin thank the continued financial support from CAPES, CNPq and FAPESP (Bergantin's Postdoctoral Fellowship FAPESP #2014/10274-3).
The authors also thank Elsevier - "author use":
Reuse of portions or extracts from the article in other works -
https://www.elsevier.com/__data/assets/pdf_file/0007/55654/AuthorUserRights.pdf.