Sarcoidosis is a granulomatous disease of unknown etiology that can affect any organ. Management of sarcoidosis patients with the potential for multi-organ involvement can be complex and require the expertise of multiple specialties. The goal of this review is to discuss clinical, imaging and epidemiologic features of organ involvement in sarcoidosis, treatment strategies and which patients are appropriate for referral to a clinic dedicated to the management of patients with sarcoidosis.
The United States Senate Resolution 443 designated April 2016 as "National Sarcoidosis Awareness Month" and pointed out some timely facts about the disease. This includes "sarcoidosis patients are often left undertreated or misdiagnosed due to the diverse presentation of sarcoidosis; the lack of knowledge of sarcoidosis among some physicians and the diagnosis of sarcoidosis through exclusion; and many sarcoidosis patients struggle to find knowledgeable physicians and emotional support resources relating to sarcoidosis; and treatment options for sarcoidosis are limited due in part to the lack of informative research and funding specific to sarcoidosis". This nicely outlines the challenges in sarcoidosis care and conveys the importance of working to improve outcomes in this underserved patient population. Primary care physicians are of vital importance in that mission. The goal of this article is to provide a succinct review of the "highlights" of the clinical presentation, imaging and management of sarcoidosis.
Sarcoidosis is a granulomatous inflammatory disease of unknown etiology that can affect any organ. Management of sarcoidosis patients with the potential for multi-organ involvement can be complex and may require the expertise of multiple specialties, but the initial recognition of the disease in its various forms often falls to Primary Care Physicians. There is substantial yearly health care cost, with an average patient cost of $18,663, and $93,201 for the top 5% per year. Sarcoidosis an area of opportunity for improving both patient outcomes and decreasing healthcare costs [1]. The prevalence of sarcoidosis is increasing in the United States, with 185,000 patients seeking out medical care annually [1]. There is significant regional variation in the prevalence of disease, with the highest rates occurring in the Northeast, South and Midwest. In the U.S., sarcoidosis is more common in females and African Americans, but can be seen in all racial and ethnic groups. There tends to be two peaks of incidence, one at about age 30 and another at about age 65. Fifty five percent of patient diagnosed with sarcoidosis are over 55 years of age at the time of diagnosis [2].
The cause of sarcoidosis is not well delineated. It is postulated that there is an environmental trigger of the disease, which results in granulomatous inflammation with a predominance of T-cell helper lymphocytes [3]. There is increasing evidence that genetics play an important role in the pathogenesis, and certain HLA subtypes are associated with disease risk as well as clinical phenotype. In pulmonary sarcoidosis, T-cell lymphocytic alveolitis precedes the histologic finding of non-necrotizing or non-caseating granulomas (5-10% may show areas of focal necrosis) [4].
Sarcoidosis is a diagnosis of exclusion and requires both a consistent clinical presentation and histologic confirmation of granulomatous inflammation. In pulmonary sarcoidosis, the use of endobronchial ultrasound-guided transbronchial needle aspiration is most commonly performed, but a thorough evaluation of other organ involvement is important and can lead to a less invasive option for biopsy. Extrathoracic lymph nodes and skin lesions are common biopsy sites if these organs are involved. Other diseases which need to be excluded include malignancy (lymphoma), hypersensitivity pneumonitis and berylliosis (excluded by history, beryllium exposure is most commonly reported in the aviation, electronics and nuclear industry). Infectious diseases can also mimic sarcoidosis, including mycobacteria tuberculosis, atypical mycobacteria, histoplasmosis, and coccidomycosis and should be excluded with fungal and AFB cultures at the time of biopsy. Certain lab tests can help support the diagnosis in combination with other findings, but should never be used in isolation to diagnose sarcoidosis. Traditional labs including angiotensin converting enzyme levels, lysozyme, and ESR have largely been replaced by soluble IL-2R and chitotriosidase levels. The association between cancer and sarcoidosis is known, studied and discussed. It involves the simultaneous or deferred presence of both entities and is mainly described in patients with lymphoma, breast cancer, melanoma, lung cancer and testicular cancer. Both pathologies require different therapeutic management, so an accurate diagnosis is necessary. On the other hand, after chemotherapy treatment, the sarcoid-like reaction of histology similar to sarcoidosis can be seen, present in draining and/or distant lymph nodes [5].
There are two clinical presentations of pulmonary sarcoidosis in which a biopsy may not be required for diagnosis. These include asymptomatic hilar adenopathy and Lofgren's syndrome. In patients presenting with asymptomatic bilateral hilar adenopathy, the statistical likelihood of sarcoidosis is so high (> 99%) that biopsy is often not pursued unless there are other findings to suggest malignancy or tuberculosis [6]. Lofgren's syndrome is a triad of erythema nodosum (painful nodular panniculitis and vasculitis), bilateral hilar adenopathy and arthralgias [7]. This presentation is more common in Caucasian patients and consists of up to 35% of cases of sarcoidosis. Fortunately, this syndrome portends an excellent prognosis, is treated with NSAIDs, and generally remits spontaneously.
Lung disease occurs in over 90% of sarcoidosis patients and is the most frequent manifestation of the disease [8]. However, clinical presentations can vary widely and can result from direct effects of sarcoidosis organ involvement or from generalized inflammation. Nonspecific findings in sarcoidosis patients include the symptoms of fatigue, or generalized pain. Pulmonary symptoms include nonproductive cough and exertional dyspnea, and patients often report wheezing. Laboratory abnormalities are also seen such as hypergammaglobulinemia and lymphocytopenia (involvement in the bone marrow) and elevations in nonspecific inflammatory markers such as sedimentation rate and CRP.
The assessment of the degree of pulmonary involvement includes symptom assessment; Pulmonary Function Tests (PFTs), and chest imaging such as a chest x-ray or CT scan (Figure 1). Pulmonary function tests in patients with sarcoidosis may show a restrictive or obstructive pattern, with obstruction usually resulting from pulmonary fibrosis and/or endobronchial sarcoidosis. The most frequent PFT abnormality noted in sarcoidosis patients is a reduced diffusing capacity, which can be due to parenchymal involvement or pulmonary hypertension, which should be considered in sarcoidosis patients who have clinical findings consistent with heart failure.
 Figure 1: Cardiac MRI, avid delayed contrast enhancement. View Figure 1
Figure 1: Cardiac MRI, avid delayed contrast enhancement. View Figure 1
Intrathoracic lymphadenopathy occurs in approximately 75% to 90% of patients, and the typical chest x-ray appearance of pulmonary sarcoidosis is that of symmetric bilateral hilar and/or paratracheal adenopathy. The most frequent parenchymal pattern on chest imaging is reticulonodular and fine linear densities and small irregular nodules, which can be more easily detected on chest CT. The Scadding stages of pulmonary sarcoidosis are based on chest x-rays and are based on the presence or absence of adenopathy, parenchymal involvement and pulmonary fibrosis. Pulmonary fibrosis is typically seen in a symmetric, upper lobe distribution. It is important to note that the prognostic value of Scadding Stages refers to the likelihood of resolution of radiographic abnormalities alone and should not be interpreted in a similar manner as cancer staging. Many patients with stage IV sarcoidosis can be asymptomatic and have a normal life expectancy. Pleural effusions and hemoptysis are infrequent [3].
In regards to extrapulmonary involvement, we feel particular attention should be paid to systems that are considered "high risk" when affected by sarcoidosis, including the eyes, heart, central nervous system, and hypercalcemia. Sarcoidosis involving these organs can lead to significant morbidity (loss of organ function) and potentially mortality (e.g., heart and CNS) if unrecognized or left untreated.
In systemic sarcoidosis, any part of the eye can be involved. Thirty to sixty percent of patients with known sarcoidosis have ocular involvement at some point in the disease. Granulomatous uveitis is the most common manifestation and can be the presenting symptom in 20-30% of patients [9]. Uveitis can result in blurred vision, watery eyes and photophobia. It is also seen as part of Heerfordt's syndrome, which may also include facial nerve palsy, parotid gland enlargement and low grade fever [10]. Other ocular manifestations include iris nodules, conjunctivitis and lacrimal gland involvement. Special attention should be paid to patients complaining of vision/visual field loss, as this can be a presenting symptom of optic neuritis which can lead to rapid vision loss if left untreated.
With the exception of the lacrimal gland and conjunctiva, biopsy of intraocular structures is generally avoided, so investigation should be undertaken of alternative sites for biopsy. Chest CT has been shown to have the highest yield for showing evidence of systemic sarcoidosis in patients presenting with eye involvement [11].
Autopsy analyses show that myocardial granulomas can be found in 20% to 30% of patients [12]. In patients with documented pulmonary sarcoidosis and no previously diagnosed cardiac sarcoidosis, one third had abnormalities on cardiac MRI and up to 80% had metabolic abnormalities on PET scanning [13]. However, the minority of patients with systemic sarcoidosis have clinical manifestations of cardiac sarcoidosis (2-3%) suggesting cardiac involvement may be under-recognized [8]. Presentations of cardiac disease include: arrhythmias including sudden death, heart block, congestive cardiac failure, angina pectoris and ventricular aneurysm formation. The Heart Rhythm Society (based on the Delphi method) recommends advanced imaging (cardiac PET or MRI) in any patient with any of the following: significant symptoms (palpitations lasting more than 2 weeks, syncope or presyncope), abnormal ECG and/or abnormal echocardiogram.
Biopsy of the heart tends to be problematic in sarcoidosis, due to the risk of the procedure and patchy nature of the disease in the myocardium, so advanced imaging is usually the diagnostic test of choice to establish a diagnosis. Cardiac MRI and cardiac PET/CT both have their advantages and disadvantages. Although there is still insufficient literature for PET/CT to be described as an evidence-based indication, on the basis of a cumulated reported accuracy (> 85%) and expert opinion that sarcoidosis is an indications for PET/CT [13] (Figure 2). Cardiac MRI (Figure 3) can reveal delayed enhancement, which pathologically is consistent with areas of scarring resulting from sarcoidosis, but can also be seen in patients with a history of myocardial infarction. This modality also gives an idea of cardiac function, and generally has a lower sensitivity but higher specificity when compared to Cardiac PET/CT scanning. Cardiac PET/CT scanning is more sensitive but less specific than cardiac MRI, but is also prone to a higher degree of false positive results [14]. A rigorous fasting protocol is required of the patients. The major advantage of cardiac PET/CT is that it also supplies information about the inflammatory activity in other areas and may also identify occult sites of involvement.
 Figure 2: Cardiac PET/CT, increased metabolic uptake in the inferolateral basal myocardium. View Figure 2
Figure 2: Cardiac PET/CT, increased metabolic uptake in the inferolateral basal myocardium. View Figure 2
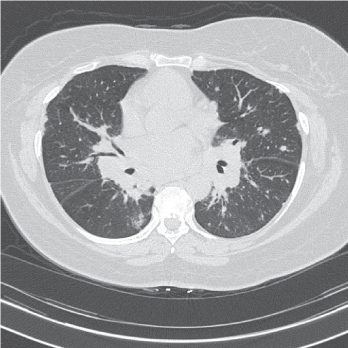 Figure 3: High resolution CT scan, innumerable pulmonary nodules (subpleural, perifissural and peribronchovascular) with bulky mediastinal and hilar lymphadenopathy. View Figure 3
Figure 3: High resolution CT scan, innumerable pulmonary nodules (subpleural, perifissural and peribronchovascular) with bulky mediastinal and hilar lymphadenopathy. View Figure 3
Upon autopsy, the central nervous system is involved in up to 25% of patients with sarcoidosis, [15] but only about 5% have symptomatic neurosarcoidosis [8]. Peripheral facial nerve palsy is the most common neurologic manifestation [15]. Other manifestations include seizures, aseptic meningitis, hydrocephalus, peripheral neuropathy and myopathy. Patients can also have spinal cord involvement, which can present with focal weakness and even paraplegia. Brain MRI with and without contrast is the diagnostic test of choice and most commonly reveals leptomeningeal enhancement. Patients can occasionally have mass-like lesions that are amenable to biopsy, but a diagnosis of presumed neurosarcoidosis can be made in the presence a clinical presentation consistent with inflammation of the meninges in combination with characteristic MRI findings in a patient with histologic evidence of sarcoidosis in another organ. Cerebrospinal fluid usually reveals nonspecific lymphocytic inflammation. The diagnostic value of measuring angiotensin converting enzyme levels in cerebrospinal fluid is controversial, and oligoclonal immunoglobulin bands can be seen, which make it difficult to differentiate sarcoidosis from multiple sclerosis [15].
Small fiber neuropathy is considered a form of neurosarcoidosis that presents a unique challenge. It can occur in up to 40% of patients with sarcoidosis [16]. Patients can present with pain or dysautonomia, and can be mistaken for fibromyalgia. This sarcoidosis-associated neuropathy involves both myelinated and nonmyelinated small nerve fibers. Diagnosis can be difficult and may require epidermal nerve fiber density testing for confirmation.
Sarcoidosis-Associated Hypercalcemia (SAHC) is seen in 10% of patients [8]. Inflammatory cytokines increase 1-alpha hydroxylase enzyme activity in the alveolar macrophages and monocytes, which leads to increased levels of the active form of Vitamin D and ultimately increased calcium absorption. Hypercalciuria is the most common measurable defect of calcium metabolism, with a prevalence of 50-62% [17]. Patients will usually present with nephrolithiasis and renal insufficiency, but can also present with nephrocalcinosis. If left untreated, SAHC can result in permanent loss of renal function. There is a high prevalence of Vitamin D deficiency (as measured by 25-hydroxy Vitamin D levels) in sarcoidosis patients, and screening for Vitamin D dysregulation in sarcoidosis should be completed prior to starting patients on Vitamin D replacement therapy, since some studies suggest Vitamin D replacement can drive disease activity in these patients. The diagnosis of SAHC or Vitamin D dysregulation in sarcoidosis requires a low/low-normal 25-hydroxy Vitamin D, high-normal/elevated 1,25-dihydroxy Vitamin D, and low/normal PTH levels in the setting of hypercalcemia or Hypercalciuria [18]. In sarcoidosis patients with Vitamin D deficiency and no evidence of SAHC, Vitamin D supplementation is safe, although recent data suggests that a lower 25-hydroxy vitamin D goal (10-20) may be more appropriate and is associated with decreased fracture risk [19].
Certain other organ manifestations of sarcoidosis are important to be aware of because of their increased prevalence and their potential for assisting in diagnosis and disease monitoring. Skin findings are seen in approximately 25% of patients with sarcoidosis [20]. Usually they occur in concert with systemic involvement, but in some cases they may be the only manifestations of the disease. Approximately 1/3 of patients with cutaneous sarcoidosis have no involvement of other organs. The skin lesions that occur may be non-specific or of 5 specific types. When biopsied, only specific skin lesions will show granulomatous inflammation. The nonspecific types of skin involvement, including erythema nodosum, do not show granulomas on biopsy. These lesions consist of tender bumps mostly on the shins and are most commonly seen in Caucasians. Calcinosis cutis, deposition of calcium salts within the skin, have also been noted in sarcoidosis and are often related to concomitant hypercalcemia [21].
Specific skin lesions show granulomas on histology. These include lupus pernio (large bluish-red and dusky purple infiltrated nodules and plaque-like lesions generally on the face and nose but can also be seen on the extremities), skin plaques, maculopapular eruptions or subcutaneous nodules. Infiltration (thickening) of old scars or tattoos (scar sarcoidosis) can be a clue to the diagnosis and can serve as reliable markers of disease activity [22]. All patients should be asked specifically about changes in existing scars or tattoos, as they often will only report these lesions when prompted.
Liver disease occurs commonly in sarcoidosis, occurring in up to 80% of patients at autopsy, however most patients with liver involvement are asymptomatic [23]. Hepatomegaly, mildly abnormal liver enzymes (usually isolated elevation in alkaline phosphatase), CT scan abnormalities (hypoattenuating nodules) and nonspecific symptoms (abdominal pain, fevers, weight loss) can be seen. Liver involvement of sarcoidosis rarely progresses to cirrhosis, portal hypertension or variceal bleeding. The majority of patients with sarcoidosis liver disease do not require therapy; however, biopsy is sometimes needed to confirm the diagnosis. Risk factors associated with hepatic sarcoidosis include African American ethnicity, prior exposure to interferon, and the presence of splenomegaly [23]. Granulomatous involvement of the spleen, abdominal lymph nodes and bones has also been noted.
Fatigue is a common symptom in sarcoidosis and has been reported in up to 50-70% of sarcoidosis patients [24]. The suspected cause is multifactorial and can include the underlying disease activity, medications and other comorbidities. Prednisone can cause significant fatigue, especially in those receiving > 500 mg/year [25]. Psychiatric comorbidities (depression/anxiety) and sleep apnea are common contributors to fatigue and should be screened for in fatigued sarcoidosis patients. When patients with stable sarcoidosis have no other identifiable cause of fatigue, medications such as Armodafinil can result in meaningful improvements in fatigue levels [26].
Disease resolution is seen in the majority of patients with sarcoidosis. Spontaneous remission was observed in 62.2% in one study, and remission or improvement following treatment was seen in another 18.7% [27]. Adverse prognostic factors include involvement of > 2 organs, age of onset < 40 yrs, and specific organ involvement (chronic hypercalcemia, lupus pernio, chronic uveitis, nephrocalcinosis, progressive pulmonary sarcoidosis, nasal mucosal involvement, cystic bone lesions, neurosarcoidosis and myocardial involvement) [28].
Chronic or progressive sarcoidosis is seen in 10-30% of patients [29]. Fewer than 5% of patients with sarcoidosis die from complications of sarcoidosis [28]. Although 80% are likely to improve over the first two years of disease activity, [30] persistent disease beyond that time is unlikely to resolve. It should be noted that treatment of sarcoidosis does not increase the likelihood of disease resolution, and is only indicated for two reasons: symptomatic management and treatment of high risk organ involvement. Decisions regarding specific medications and doses should be made based which sarcoidosis medication is the primary driver of symptoms or risk. Currently, there are only two FDA approved drugs for the treatment of pulmonary sarcoidosis: Prednisone and repository corticotropin injection. Prednisone is considered first line therapy for most forms of sarcoidosis. For pulmonary sarcoidosis, 20-40 mg daily is considered adequate initial dosing, with the goal of tapering to the lowest effective dose over 3-6 months, which is then continued for 6-12 months. Cardiac or neurologic sarcoidosis usually requires higher initial doses of steroids. Symptom recurrence or disease progression as the dose is tapered should lead to consideration of second-line therapy. It should be remembered that the primary goal of second line medications is to serve as a means of allowing steroids to be tapered without disease progression or recurrence. Repository corticotropin injection has received relatively little attention to date and lacks robust research to support its use as anything other than a third or fourth line medication [31].
Several studies have demonstrated the value of methotrexate in patients with refractory disease, and it generally considered the initial choice second-line medication [32]. This medication is generally given orally or subcutaneously weekly. Daily folic acid supplementation is recommended by most experts to prevent folate depletion and potential side effects such as oral ulcers and hair loss. Contraindications to methotrexate include pre-existing liver or renal pathology. In Dr. Baughman's seminal study in 1995, improvements in vital capacity or other affected symptomatic organ was noted in 33 of 50 patients treated with methotrexate and corticosteroids were discontinued in an additional six patients who remained stable with clinical or symptomatic improvement [33]. The major toxic effects noted in 150 patient-years of therapy were hepatic, leukopenia requiring hospitalization, and cough. In our experience, methotrexate is a well-tolerated and effective steroid sparing medication in sarcoidosis, and should be considered the first-line steroid sparing therapy. Unfortunately, the half-life of methotrexate is long, so it can take from 6 weeks to 6 months to take full effect. Overlapping therapy with prednisone is usually required, and we often delay weaning of prednisone until patients have been on methotrexate for at least 6 weeks. In another study, two European medical centers were compared where one used methotrexate as a second-line anti-sarcoidosis agent and the other used azathioprine [34]. The rate of drug side effects, particularly the rate of infection, was significantly greater in the patients receiving azathioprine as a second-line agent, although there was no difference in the reduction in corticosteroid dose or improvement of pulmonary function.
The Tumor Necrosis Factor Alpha (TNF-α) antagonistinfliximab appears to be an effective third line medications in sarcoidosis and is often used if patients have intolerance or therapeutic failure of methotrexate or azathioprine. A retrospective study indicates that long-term infliximab is very efficient and safe in patients with chronic steroid-resistant sarcoidosis and patients with predominantly extrapulmonary sarcoidosis seem to get significant benefit as well [35]. This medication is generally well tolerated, however its high cost, and fact that it is not FDA approved for the treatment of sarcoidosis limits treatment options. In our experience in the United States, insurance coverage can be procured if intolerance or ineffectiveness of methotrexate or other second line medications are documented. Infliximab results in improvement in skin lesions, pulmonary function and inflammatory markers, including PET scan and neurological improvement in neurosarcoidosis patients [36,37]. IVIG has also been used as therapy in refractory neurosarcoidosis [16].
Sarcoidosis patients are often on one or more immunomodulating therapies which lead itself to the need for close care coordination. Multiple referrals to different specialties and disciplines are often necessary. A difficult question is when to refer patients to a specialized center. A minority of academic medical centers have dedicated sarcoidosis clinics, and the existence of a dedicated sarcoidosis clinic is associated with the number of sarcoidosis patients seen annually [38]. As an orphan disease there is variability in sarcoidosis practice patterns between centers, and this is also evidence in the organizational structure of dedicated sarcoidosis clinics. The minority of dedicated sarcoidosis clinics used a concurrent multidisciplinary model [38]. As sarcoidosis is a systemic disease that can affect any organ system as outline above, [39] it is somewhat intuitive that a multispecialty clinic model could improve patient experience and outcomes. Rheumatology clearly plays an important role in the multidisciplinary management of sarcoidosis [40]. The most common other subspecialties other than pulmonary that are involved in the currently existing multispecialty clinics include those associated with the more common disease manifestations (dermatology and ophthalmology) and the manifestations that can be life threatening (cardiology and neurology) [41]. A multidisciplinary model for evaluation, diagnosis, and treatment could lead to more reliable conclusions and progress in sarcoidosis through collaboration and data sharing. Such models have proven to improve outcomes in other diseases, including cystic fibrosis, [42] and hopefully will exist in the future for patients affected with sarcoidosis. However, primary care physicians will remain on the front lines of diagnosis for the majority of sarcoidosis patients, and are the most important providers for initiating effect disease management.