Thrombotic microangiopathy is a group of syndromes characterized by thrombocytopenia, microangiopathic hemolytic anemia, and acute organ dysfunction secondary to ischemia. It includes a group of diseases such as thrombotic thrombocytopenic purpura. They can be primary or secondary to multiple pathologies, including autoimmune diseases such as systemic lupus erythematosus. These associations are important from the clinical point of view, as they have more severe presentations, high relapse rates, and in some cases poor response to conventional treatment. We present the case of a patient with thrombotic microangiopathy associated with SLE with characteristics of a classic thrombotic purple thrombocytopenia. Combination therapy was treated, achieving an adequate clinical response.
Thrombotic microangiopathy, Thrombotic thrombocytopenia Purpura, Systemic lupus erythematosus, Plasmapheresis, Rituximab
Thrombotic microangiopathy (TMA) is a group of syndromes that includes Thrombotic thrombocytopenic purpura (TTP), which is conditioned by a deficiency of the metalloproteinase ADAMTS13 (a metalloproteinase similar to disintegrin and with thrombospondin type 1 motif # 13); It can be primary or secondary, and within etiologies of the latter, autoimmune diseases such as Systemic lupus erythematosus (SLE) [1,2].
We present the case of a patient with systemic lupus erythematosus presenting with thrombotic thrombocytopenic purpura.
Female patient, 38-years-old, with a 7-day medical history with generalized abdominal pain associated with nausea and jaundice, without other associated symptoms. The patient has a history of SLE diagnosed at 28-years-old, with joint, hematological (Leukopenia) and skin deterioration (Raynaud's phenomenon), she was being treated with chloroquine, low-dose prednisone, nifedipine, and methotrexate.
On admission, pancytopenia is documented; lymphopenia, non-immune macrocytic hemolytic anemia (indirect hyperbilirubinemia, Lactic dehydrogenase (DHL) and elevated reticulocytes, consumed haptoglobin and direct negative coombs), severe thrombocytopenia, associated with proteinuria (Table 1). Considering initially the lupus activity, treatment with pulses of methylprednisolone was started, in addition, methotrexate was interrupted due to suspicion of myelotoxicity.
Table 1: Admission laboratories. View Table 1
During his hospitalization, schistocytes (1+) were documented in a Peripheral blood smear (ESP), however, in the context of microangiopathic hemolytic anemia and thrombocytopenia, TMA was considered as an alternative diagnosis, starting with plasmapheresis and cyclophosphamide, expanding studies with ADAMTS13 measurement.
Studies were concluded, excluding infectious etiologies (negative hepatotropes, cytomegalovirus, human immunodeficiency virus and dengue), deficiency (normal folic acid and vitamin b12) and metabolic (normal liver and thyroid function). Studies with elevated acute phase reactants, complement consumption and negative anti-DNA.
The referred treatment was started, presenting generalized tonic-clonic seizures during admission, extension studies ruled out the presence of an acute cerebrovascular event (simple skull tomography and normal brain angio-resonance), and there were no alterations in videotelemetry. The report of ADAMTS13 with 0% activity was received, compatible with PTT, considering the initiation of rituximab in a medical meeting to be beneficial, based on the high rate of recurrences in this type of patients, started with a dose of 375 mg/m2 per week and 7 sessions of plasmapheresis were performed.
The patient presented a favorable response to the established treatment, reaching normalization of the platelet count and DHL (see Figure 1), she was discharged with antimalarial, oral corticosteroid at a dose of 1 mg/kg and remaining doses of anti-CD20.
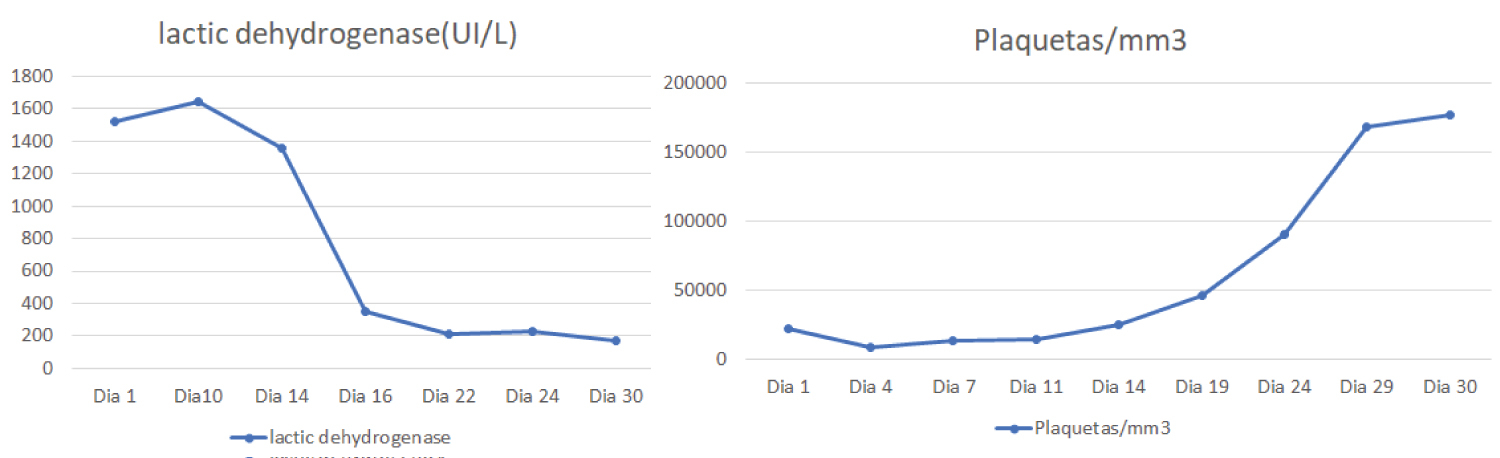 Figure 1: Behavior of lactic dehydrogenase and platelets. View Figure 1
Figure 1: Behavior of lactic dehydrogenase and platelets. View Figure 1
TMA is a syndrome characterized by microangiopathic hemolytic anemia associated with thrombocytopenia and ischemic injury secondary to arteriolar and capillary thrombosis of different organs. It is a spectrum of diseases: TTP, atypical hemolytic-uremic syndrome and hemolytic-uremic syndrome [3].
PTT can be classified into primary and secondary. The first group includes presentations due to decreased activity of ADAMTS13 due to mutations in its gene. In the secondary group, of which our patient is a part, the cause is more related to the presence of antibodies or to complement mediation and does not include only autoimmune pathologies, also neoplastic, infectious, related to pregnancy, medications, metabolic disorders and post-transplant [4-6].
TMAs associated with autoimmune diseases can be classified as shown in (Figure 2) [4].
 Figure 2: TMA classification associated with autoimmune diseases [4].
Figure 2: TMA classification associated with autoimmune diseases [4].
*TMA: Thrombotic microangiopathy; TTP: Thrombotic thrombocytopenic purpura; APS: Antiphospholipid syndrome
View Figure 2
The prevalence of SLE and TTP is approximately 1 to 4%, generally more observed in young black women and although mortality is low in classic TTP, when it is associated with SLE the picture is different, reaching between 34 and 62.5%, also associated with higher recurrence rates [7-9].
Generally, TTP associated with SLE does not decrease the activity of ADAMTS13, other mechanisms were found such as resistance to von Willebrand factor proteolysis due to the pro-inflammatory environment of the disease, the presence of anti-endothelial antibodies and complement activation [9].
SLE-associated TTP should be considered when microangiopathic hemolytic anemia or thrombocytopenia is more severe than expected for the underlying disease or severe neurological and/or kidney disorders. They tend to present aggressive renal failure, even more so in the presence of antiphospholipid antibodies with severe proteinuria, in most cases there is no association with disease activity and is sometimes limited to the kidney [4].
As risk factors for mortality in TTP and patients with SLE, Kwok, et al. [10] found the presence of a high SLEDAI score (> 10 and neurological manifestations points) (systemic lupus erythematosus disease activity index) and coexisting nephritis, in addition to infections. Other risk factors identified are lymphopenia and hemoglobin levels below 7 g/dL [11]. These data are similar to those published by Jian, et al. [12], finding that class IV lupus nephritis and renal thrombotic microangiopathy are also associated with higher mortality. Other findings of this study are that up to 40.6% of the patients showed a < 5% decrease in ADAMTS13 activity, similar to what happened in our case.
Regarding the treatment of this type of patients, there are several therapies used, such as plasmapheresis, corticosteroids, cytotoxic drugs, biological therapy and immunoglobulin. TMA associated with rheumatological disease has more relapses, so that therapies combined with cytotoxic or biological therapy have a higher remission rate than plasmapheresis or isolated immunosuppressants [8,13,14].
Regarding refractory cases or those with a high risk of recurrence, rituximab has shown encouraging clinical results, which is why it was chosen for our patient, obtaining an adequate response, with no recurrence so far [8]. Eculizumab, a monoclonal antibody that inhibits C5 activation, can potentially be used in refractory cases [15].
In conclusion, it is important to consider TMA as a differential diagnosis in patients with rheumatological diseases, given its high mortality and recurrence, and to establish early combined therapy, as in our case.
None.
The authors declare that they have no conflict of interest.