Rilpivirine (RPV) is a second-generation non-nucleoside reverse-transcriptase inhibitor used in combination antiretroviral therapy (cART) in naive and experienced HIV-positive adult subjects. To evaluate its efficacy and safety in these patient settings, we performed a meta-analysis of randomized controlled trials with available data at 48 and 96 weeks of follow-up. We considered 4 studies involving 2336 cART-naïve patients and 8 studies involving 3165 cART-experienced virologically controlled patients. Regarding efficacy, the virological response rate and the mean difference in the change from the baseline CD4 cell count were not significantly different between the RPV and comparator arms in both patient groups at both time points. Regarding safety, the discontinuation rates due to any adverse events (AEs), serious AEs, RPV-related AEs and AEs leading to drug discontinuation did not significantly differ from the rates in the comparator group at both time points. A systematic review of lipid changes was also performed: the safety and advantageous metabolic impact of RPV on lipids, especially among cART-naïve subjects at up to 96 weeks of follow-up, were confirmed. Our meta-analysis indicated that RPV-based regimens were effective and tolerable for both types of patients, which was consistent with published data from real-life settings.
Rilpivirine (RPV; TMC278; Edurant®) is a second-generation non-nucleoside reverse-transcriptase inhibitor (NNRTI) with activity against many viral strains resistant to previous NNRTIs and a moderate-high genetic barrier to resistance development [1,2]. RPV efficacy and safety have been assessed in registrative randomized controlled clinical trials (RCTs) in HIV-positive treatment-naïve [3-7] and treatment-experienced patients [8-14] with documented long-term efficacy and tolerability. Real-life data from observational studies [15-21] eventually confirmed these results. Therefore, current Italian [22], European [23], British [24,25] and DHHS (Department of Health and Human Services) [26] HIV/AIDS guidelines recommend the use of RPV as a first-line third agent coupled with a nucleoside reverse transcriptase inhibitor backbone in people living with HIV (PLWH) with CD4 count > 200 cells/μL and HIV RNA < 100,000 copies/mL starting combination antiretroviral therapy (cART) and in optimization strategies represented by RPV-based single tablet regimens (both standard three-drug-based cART and, with some restrictions, dual regimens combined with dolutegravir). Furthermore, susceptibility to RPV is not affected by the presence of single common NNRTI resistance-associated substitutions, so it retains good antiviral activity against K103N and other codon-mutated strains [1,2,27]. Lastly, long-acting injectable formulations allowing once-monthly or more distanced dosing with RPV sustained-release are under investigation (NCT03299049, NCT02938520, NCT03639311, and NCT02951052) or have already shown promising results, [28,29] being ready to be widely prescribed in clinical practice.
RPV is administered orally as a 25-mg tablet once daily with food and is available alone or as a fixed-dose combination with emtricitabine/tenofovir (disoproxil or alafenamide) or with dolutegravir. No dosage adjustment is recommended for patients with renal dysfunction or mild-to-moderate hepatic insufficiency [1,2,27]. Although RPV is metabolized by CYP3A4-mediated hepatic oxidation, no inhibition or induction of cytochrome P-450 isoenzymes has been reported, and its spectrum of interaction is favorably narrowed [1,2,27].
To date, one meta-analysis in 2014 has been performed on the efficacy and safety of RPV in treatment-naïve only adult PLWH, with efavirenz (EFV) [30] as the comparator. Considering the changing clinical background, as well as the versatility and various advantages of RPV, we conducted a systematic review and meta-analysis to evaluate the 48-week and long-term (defined as at least 96 weeks) efficacy (as the proportion of plasma HIV RNA < 50 cp/mL and as the change in CD4 cell/count) and safety (as the incidence of adverse events and as the change in lipid levels) of RPV-based cART compared to other current cART regimens in both treatment-naïve and treatment-experienced virologically suppressed PLWH. The data reporting followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines [31].
Relevant studies were identified by systematic electronic literature searches in the Medline/PubMed, EMBASE and Cochrane Library databases from inception to June 2019. No language restriction was applied. The detailed search strategy is described in the Supplementary Methods. In addition, we manually reviewed the references of recent reviews and meta-analyses and of all the papers selected for inclusion to identify additional studies that met the inclusion criteria. The flowchart with detailed information on the search and selection process is presented in Figure 1.
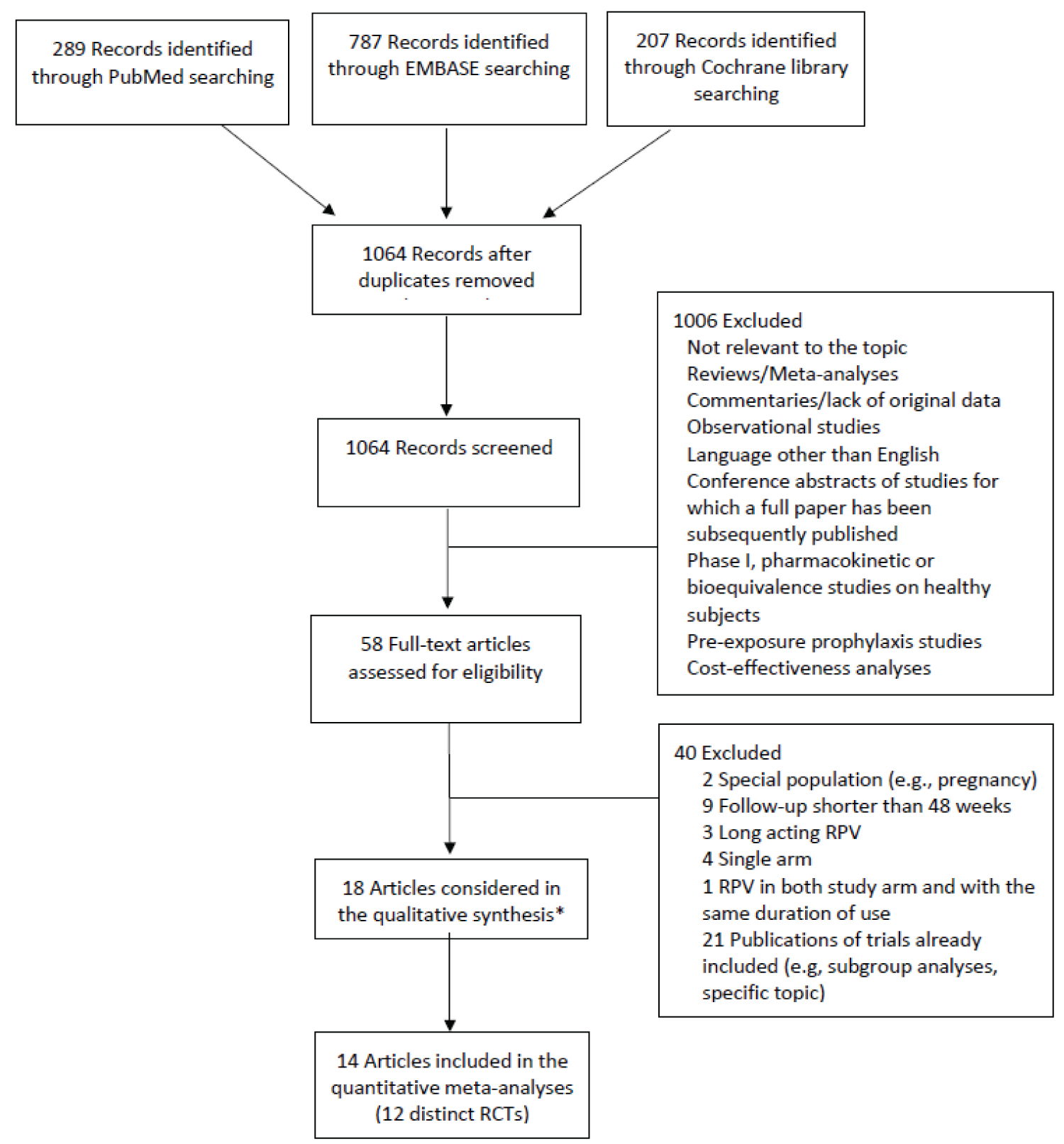 Figure 1: Flowchart describing the literature search and study selection process.
Figure 1: Flowchart describing the literature search and study selection process.
*Including two articles from which we extracted information on changes in lipid levels separately from the ECHO and THRIVE trials at 48 weeks of follow-up and 2 articles from the TMC278-C204 and SWORD 1&2 trials reporting results at 192 and 148 weeks of follow-up, respectively, that were not used in the meta-analyses.
Abbreviations: RCT: Randomized clinical trial; RPV: Rilpivirine.
View Figure 1
RCTs with at least 48 weeks of follow-up and comparing the efficacy (viral suppression) and safety of RPV use (dose: 25 mg/day) with other cART in adult PLWH were included. Observational studies, reviews, meta-analyses, commentaries without original data, phase 1 studies, pharmacokinetic or bioequivalence studies on healthy subjects, pre-exposure prophylaxis studies, cost-effectiveness analyses, and non-English-language articles were excluded. The conference abstracts of relevant RCTs were considered only if a full-length publication was not available. Single-arm studies, studies based on special populations (i.e., pregnant women), studies investigating long-acting RPV, studies in which RPV was used in both arms and for the same time period, and studies with a follow-up shorter than 48 weeks were also excluded. Studies comparing immediate to delayed switch to RPV-based regimens were considered eligible, and the delayed switch arm was used as the comparator in the analyses. In the case of multiple publications from the same trial, we selected the main trial publications with results of the primary efficacy outcome (i.e., viral suppression) at 48 and 96 (± 8) weeks of follow-up. No studies were excluded a priori for weakness of design or data quality.
The merging of the records identified from Medline/PubMed, EMBASE and the Cochrane Library resulted a total of 1064 unique records. Two reviewers independently screened the titles and abstracts of all the retrieved articles and examined the full texts of the articles considered potentially eligible for inclusion (n = 58). Any disagreements were resolved by consensus. Eighteen articles met the inclusion criteria and were included in the systematic review [3-14,28,32-36] (Table 1). Four of these articles were presented in tables and were not included in the quantitative meta-analyses of the efficacy and safety outcomes. They were the two separate publications of the ECHO [3] and THRIVE [4] trials at 48 weeks of follow-up (which were used when reporting results for lipid values, as the publication of the pooled analysis did not detail such quantitative information [36]) and the publications at 148 weeks of follow-up of the SWORD 1-2 trials [35] and at 192 weeks of the TMC278-C204 trial [6] (which were not used in the meta-analyses, as no other study had data for a comparable follow-up period). Thus, a total of 14 publications [5,7-14,28,32-34,36], which provided results on 12 distinct RCTs at 48 and/or 96 (± 8) weeks of follow-up, were included in the meta-analyses. The data were extracted from the selected articles in a standardized format by 2 independent reviewers. The data collected from each study included trial name, enrollment period, geographic area, number of patients included and treatment regimens, patient characteristics at baseline (e.g., cART-naïve or experienced patients, cut-off for plasma viral load and CD4 cell count for inclusion, age, and treatment duration before study inclusion for cART-experienced patients), duration of follow-up, and results for the efficacy and safety endpoints according to the ITT analysis. Data on viral suppression according to plasma viral load and CD4 cell count at baseline were also extracted from trials on cART-naïve patients. When available, the results of the US Food and Drug Administration (FDA) Snapshot algorithm were extracted. Our primary efficacy endpoint was the proportion of participants with viral suppression (defined as HIV RNA levels < 50 copies/mL) at 48 and 96 (± 8) weeks of follow-up. One study that defined viral suppression as HIV RNA levels < 40 copies/mL was also considered [14]. The secondary efficacy endpoint was the change in CD4 cell counts from baseline at 48 and 96 (± 8) weeks of follow-up. Safety endpoints include the number of any AEs, serious AEs, drug-related AEs and AEs leading to study discontinuation. Information on the change in lipids and glucose levels at 48 and 96 weeks of follow-up was also extracted. The TMC278-C204 study was a dose-ranging trial with three once-daily RPV doses (i.e., 25, 75 or 150 mg) [33]. In the meta-analyses, we included the results for the 25 mg dose only. The LATTE trial design included a 24-week induction phase assessing the antiviral activity and safety of 3 different cabotegravir doses over efavirenz in cART-naïve adult PLWH, followed by a 72-week maintenance phase assessing cabotegravir plus RPV (over EFV) for the maintenance of viral suppression [28]. Only results based on the maintenance-exposed population were included in the meta-analyses. That patient population was considered ART-experienced. The individual study quality assessment was performed with Version 2 of the Cochrane risk-of-bias tool for randomized trials (RoB 2) [37] in relation to the specific outcome plasma HIV RNA.
Table 1: Main characteristics of trials comparing rilpivirine (RPV)-based regimens with other antiretroviral therapies (ART). View Table 1
The meta-analyses of the efficacy outcomes (i.e., HIV RNA levels < 50 copies/mL and change in CD4 cell counts from baseline) were conducted separately for the 48-week and 96-week time points and for the treatment-naïve and treatment-experienced virologically controlled patients. In the meta-analyses of the safety outcomes, we combined all the trials and used the data at the longest follow-up. Risk ratios comparing the number of events in patients treated with RPV-based ART versus those treated with other ART were used as the effect measures for the viral suppression and AE endpoints. For the CD4 cell count endpoint (change from baseline), we used the weighted mean difference as the effect measure. Study-specific RRs (and 95% CIs) of viral suppression and AEs were calculated from the distribution of the outcomes in the RPV and comparator arms. Study-specific mean differences and 95% CIs in the change in CD4 cell count from baseline for the RPV arm vs. the comparator arm were calculated from the mean values of the change and the corresponding standard deviations, standard errors or 95% CIs in the two arms. The summary RR and mean difference were estimated through random-effects models to take into account the heterogeneity of the risk estimates, using the moment estimator of the variance across studies as the weight [38,39]. Heterogeneity among trials was evaluated using the χ2 test (defined as a p-value less than 0.10), and inconsistency was measured using the I2 statistic, i.e., the percentage of total variation attributable to among-study heterogeneity rather than chance (values of the I2 statistic of approximately 25%, 50% and 75% are indicative of low, moderate and high heterogeneity, respectively) [40]. Publication bias was evaluated through the examination of funnel plots and tests proposed by Begg and Mazumdar and Egger [41,42]. We conducted sensitivity analyses by excluding each study one by one from the meta-analysis and subgroup analyses of viral suppression according to viral load (≤ 100,000/> 100,000 copies/mL) and CD4 cell count (< 200/> 200 cells/mm3) at baseline in treatment-naïve patients. All statistical analyses were performed using STATA software (version 14.1; StataCorp, College Station, TX, USA).
A search of electronic scientific databases led to identify a total of 1064 records. After title and abstract reviewing and checking for duplicates using Endnote X7 software, we excluded 1006 records mainly because they presented data from populations or study designs not matching our inclusion criteria (i.e., healthy subjects, observational studies) or not reporting original data (i.e., reviews, commentaries, opinion letters). Fifty-eight articles on RPV were thoroughly analyzed: All the full texts were downloaded and evaluated for the inclusion in this meta-analysis. Another 40 of these articles were excluded mainly because they reported replicated data or matched one or more of our exclusion criteria. At the end of the study selection process, after excluding the last 4 articles exclusively used for extracting data on RPV safety for the qualitative synthesis of lipid changes, 14 original articles reporting on 12 distinct RCTs were eventually included and presented in the meta-analysis. A brief flow-chart of the study selection process is shown in Figure 1. Two study groups were considered based on the treatment status of the enrolled patients: cART-naïve and cART-experienced virologically suppressed subjects. Only studies with available 48 and 96 weeks data of follow-up were included to perform the meta-analysis. The characteristics of both studies design and enrolled patients included in our analysis are reported in Table 1. The main results in terms of changes in CD4 cell counts, lipids and glucose levels of the studies included in the meta-analysis are summarized in Supplementary Table 1 and Supplementary Table 2.
Supplementary Table 1: Change in CD4+ cell count in trials comparing rilpivirine-(RPV)-based regimens with other antiretroviral therapies (ART). View Supplementary Table 1
Supplementary Table 2: Changes in lipids in trials comparing rilpivirine (RPV)-based regimens with other antiretroviral therapies (ART). View Supplementary Table 2
Individual study quality assessment is depicted in Supplementary Figure 1. The study protocols were obtained where available to assess selective outcome reporting. All the studies detailed the statistical process for the outcomes and addressed missing or incomplete data (i.e. loss to follow-up); therefore, the risks of bias due to either missing outcomes, measurement of the outcome or selection of the reported results were ranked as low. They all achieved adequate sequence generation and allocation concealment, but the use of blocked randomization of size 4 in a single recruitment center may suggest minor issues with the allocation concealment in the study by Petchkum, et al. [14]. Biases related to potential deviations from the intended interventions were also deemed low for all the RCTs, except for the PROBE study [11], which did not report detailed information on patient disposition to make a judgment.
 Supplementary Figure 1: Risk of bias according to the revised Cochrane risk-of-bias tool for randomized trials (RoB 2).
View Supplementary Figure 1
Supplementary Figure 1: Risk of bias according to the revised Cochrane risk-of-bias tool for randomized trials (RoB 2).
View Supplementary Figure 1
Efficacy was primarily defined as the proportion of participants with viral suppression (defined as plasma HIVRNA levels < 50 copies/mL) at 48 and 96 weeks (± 8 weeks) for cART-naïve adult PLWH and at 48 weeks (± 8 weeks) for cART-experienced virologically controlled adult PLWH.
For cART-naïve patients, we included 4 studies in this meta-analysis (TMC278-C204, ECHO, THRIVE and STAR) covering a total of 2336 adult patients evaluated at 48 and 96 weeks of follow-up from 5 distinct publications. In the intention-to-treat (ITT) analysis, the virological response rate with RPV was not significantly different from the comparator at weeks 48 and 96, with risk ratio (RR) values equal to 1.04 (95% confidence interval (CI) 1.00-1.08) and 1.03 (95% CI 0.98-1.09), respectively. A very low degree of heterogeneity was found between the RR estimates at week 48 (I2 0.0%, p = 0.706) and week 96 (I2 19.2%, p = 0.290; Figure 2).
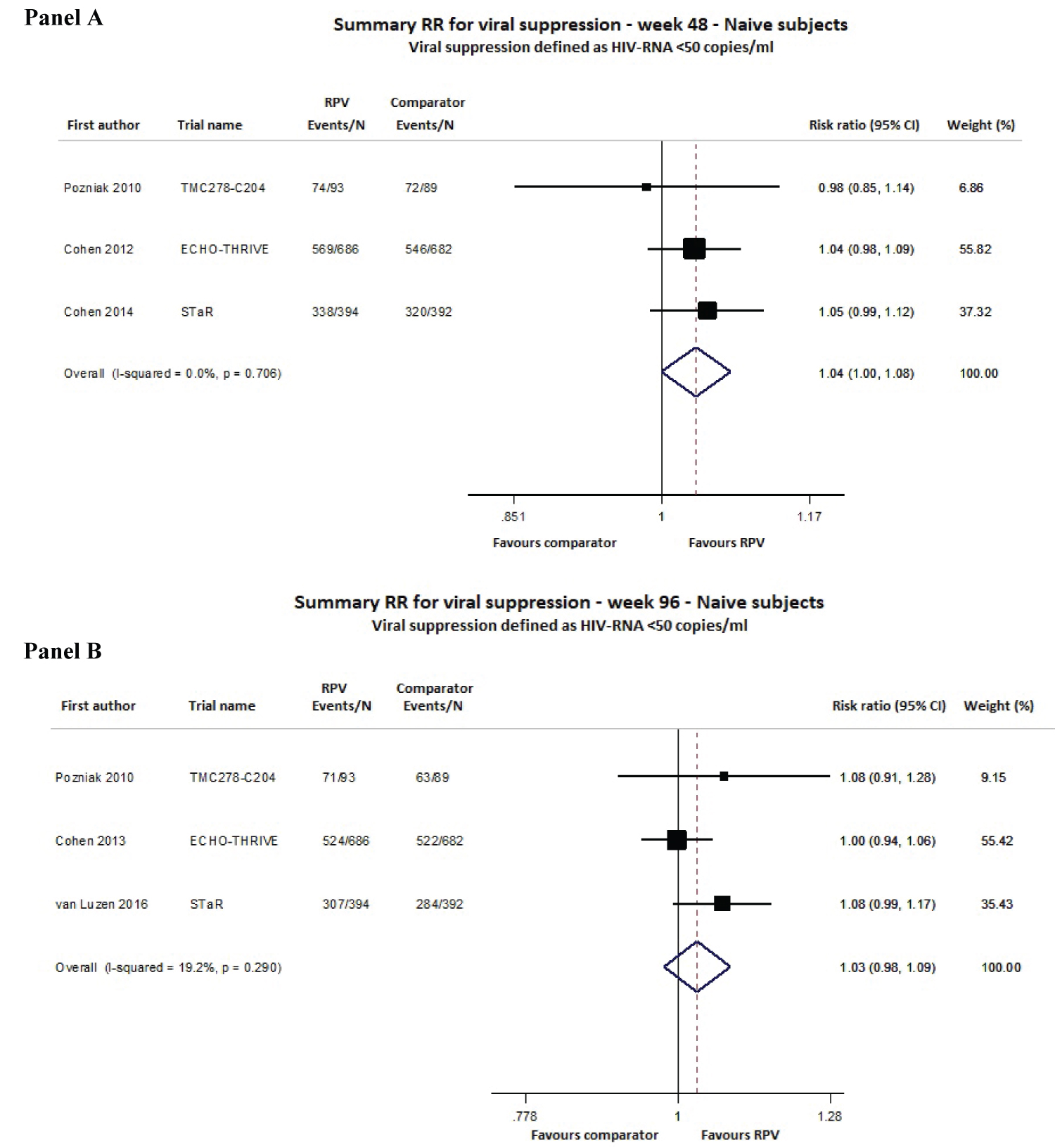 Figure 2: Meta-analysis of viral suppression (defined as HIV RNA levels < 50 copies/mL) for ART-naïve adult subjects at 48 (Panel A) and 96 (Panel B) weeks of follow-up.
View Figure 2
Figure 2: Meta-analysis of viral suppression (defined as HIV RNA levels < 50 copies/mL) for ART-naïve adult subjects at 48 (Panel A) and 96 (Panel B) weeks of follow-up.
View Figure 2
For cART-experienced virologically controlled patients, data were available from 8 studies (SPIRIT, LATTE, PROBE, GS-US-366-1160, SWORD-1, SWORD-2, SALIF and NCT03664440) from 7 publications for a total of 3165 adult PLWH. At week 48, the ITT analysis showed that the virological response rate was once again not significantly different from the comparator (RR 0.99; 95% CI 0.97-1.01), and the heterogeneity test again showed a very low variability among the studies (I2 0.0%, p = 0.736; Figure 3). No evidence of publication bias was detected for the cART-naïve or cART-experienced analyses (Supplementary Figure 2).
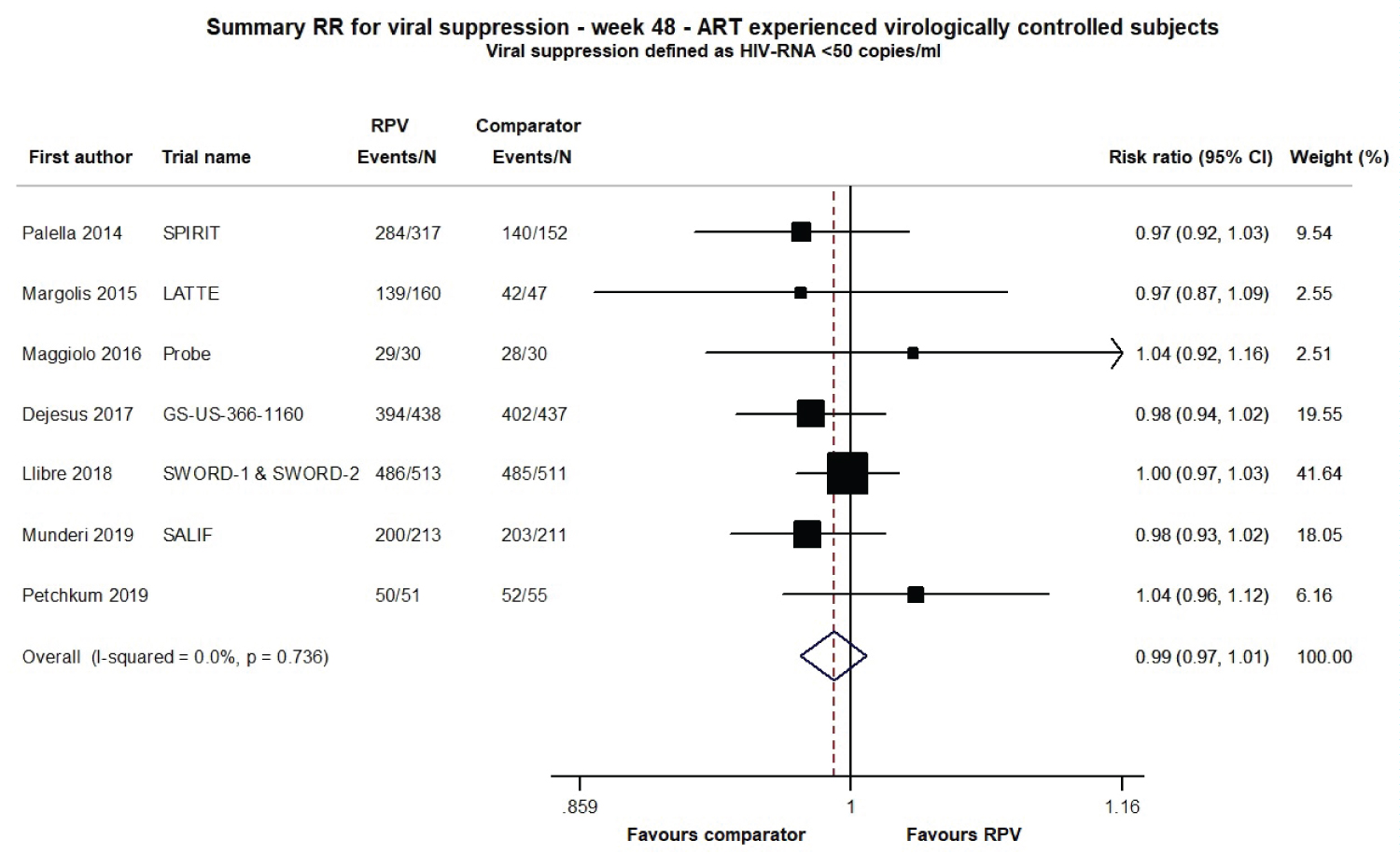 Figure 3: Meta-analysis of viral suppression (defined as HIV RNA levels < 50 copies/ml) for ART-experienced virologically controlled adult subjects at 48 weeks of follow-up.
Figure 3: Meta-analysis of viral suppression (defined as HIV RNA levels < 50 copies/ml) for ART-experienced virologically controlled adult subjects at 48 weeks of follow-up.
Note: Comparator group in the SPIRIT trial: 24 weeks of ongoing ART + 24 weeks of RPV/FTC/TDF.
View Figure 3
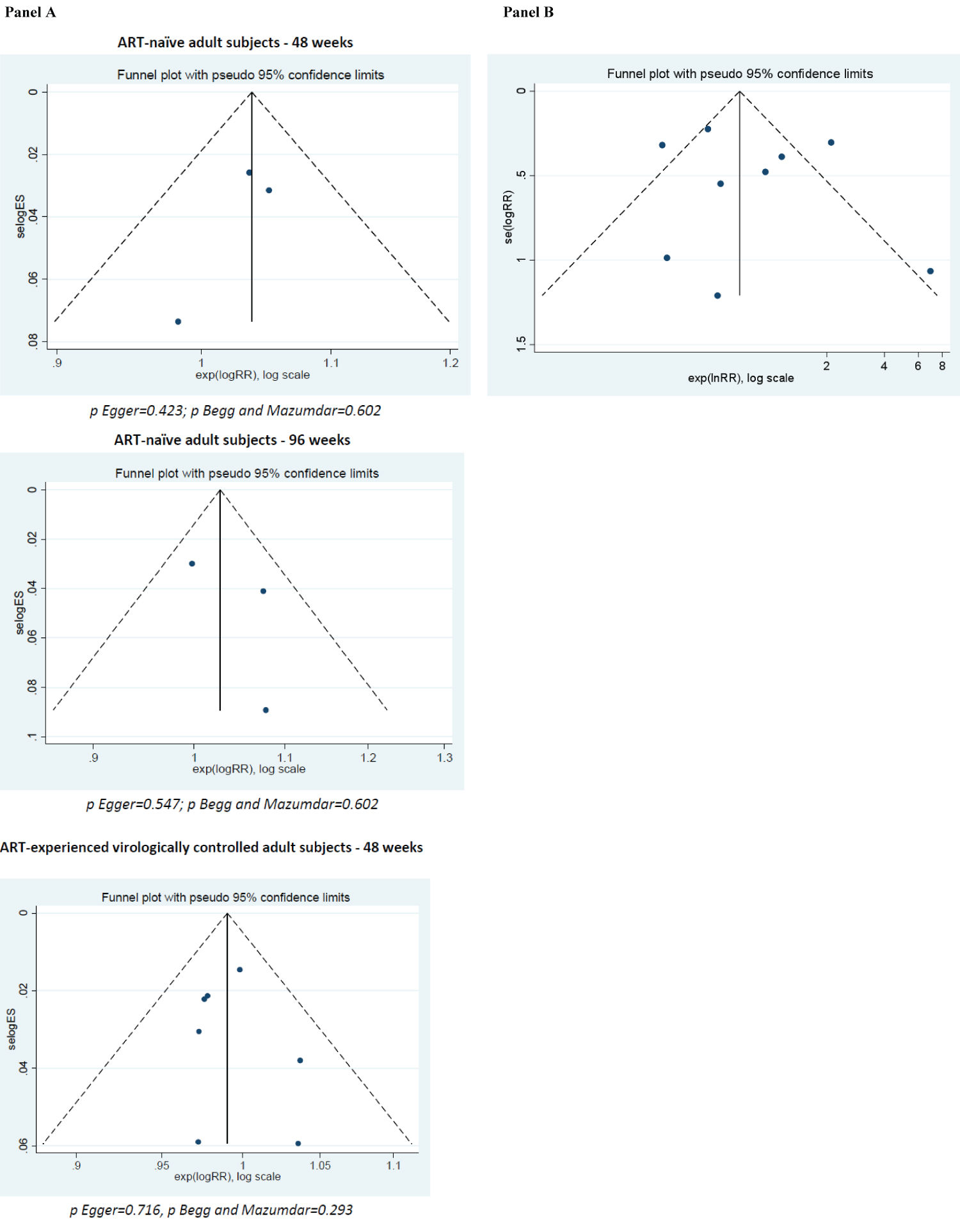 Supplementary Figure 2: Publication bias assessment (funnel plots, Egger and Begg and Mazumdar’s tests) for the plasma viral load outcome (panel A) and for the discontinuation due to adverse events outcome (panel B).
Supplementary Figure 2: Publication bias assessment (funnel plots, Egger and Begg and Mazumdar’s tests) for the plasma viral load outcome (panel A) and for the discontinuation due to adverse events outcome (panel B).
p Egger = 0.611; p Begg and Mazumdar = 0.835.
View Supplementary Figure 2
In subgroup analyses conducted in naïve subjects at weeks 48 and 96, we calculated pooled RRs according to the baseline plasma viral load (2 studies, 2154 subjects) and the baseline CD4 cell count (2 studies, 2154 subjects). The virological response rate with RPV was not significantly different from the comparator among patients starting an RPV-based regimen with baseline HIVRNA > 100,000 copies/mL either at week 48 (RR 0.96; 95% CI 0.90-1.03) or at week 96 (RR 0.96; 95% CI 0.88-1.05; data not shown). In contrast, among patients starting an RPV-based regimen with baseline HIV RNA < 100,000 copies/mL, the RPV group weakly trended toward a better virological response rate than the comparator at weeks 48 (RR 1.08; 95% CI 1.03-1.13) and 96 (RR 1.07; 95% CI 1.01-1.13; data not shown). At both follow-up times, the virological response rate in the RPV group was not significantly different from that in the comparator among patients starting cART with either a CD4 count above or below 200 cells/μL, except at week 48. In the latter case, patients with a CD4 count > 200 cells/μL showed a mild trend for a better virological response if starting RPV instead of the comparator (RR 1.06; 95% CI 1.02-1.11; data not shown). No heterogeneity was observed in any of the sub-analyses.
Efficacy was secondarily assessed in terms of the change in CD4 cell counts from baseline at 48 and 96 weeks (± 8 weeks) of follow-up for the cART-naïve adult group and at 48 weeks for the cART-experienced virologically controlled adult group.
For cART-naïve patients, data were available from 4 studies, for a total of 2336 adult PLWH evaluated at week 48 and week 96 (5 distinct publications; Supplementary Table 1). The ITT analysis showed that the mean difference (MD) in the change in CD4 cell counts from baseline to week 48 did not differ between RPV and the comparator group (MD 10.93; 95% CI -1.03-22.90; Figure 4), and a similar result was observed at week 96 (MD 8.01; 95% CI -6.94-22.95; Figure 4). The heterogeneity assessment showed no significant variability among the studies at either time point (Figure 4).
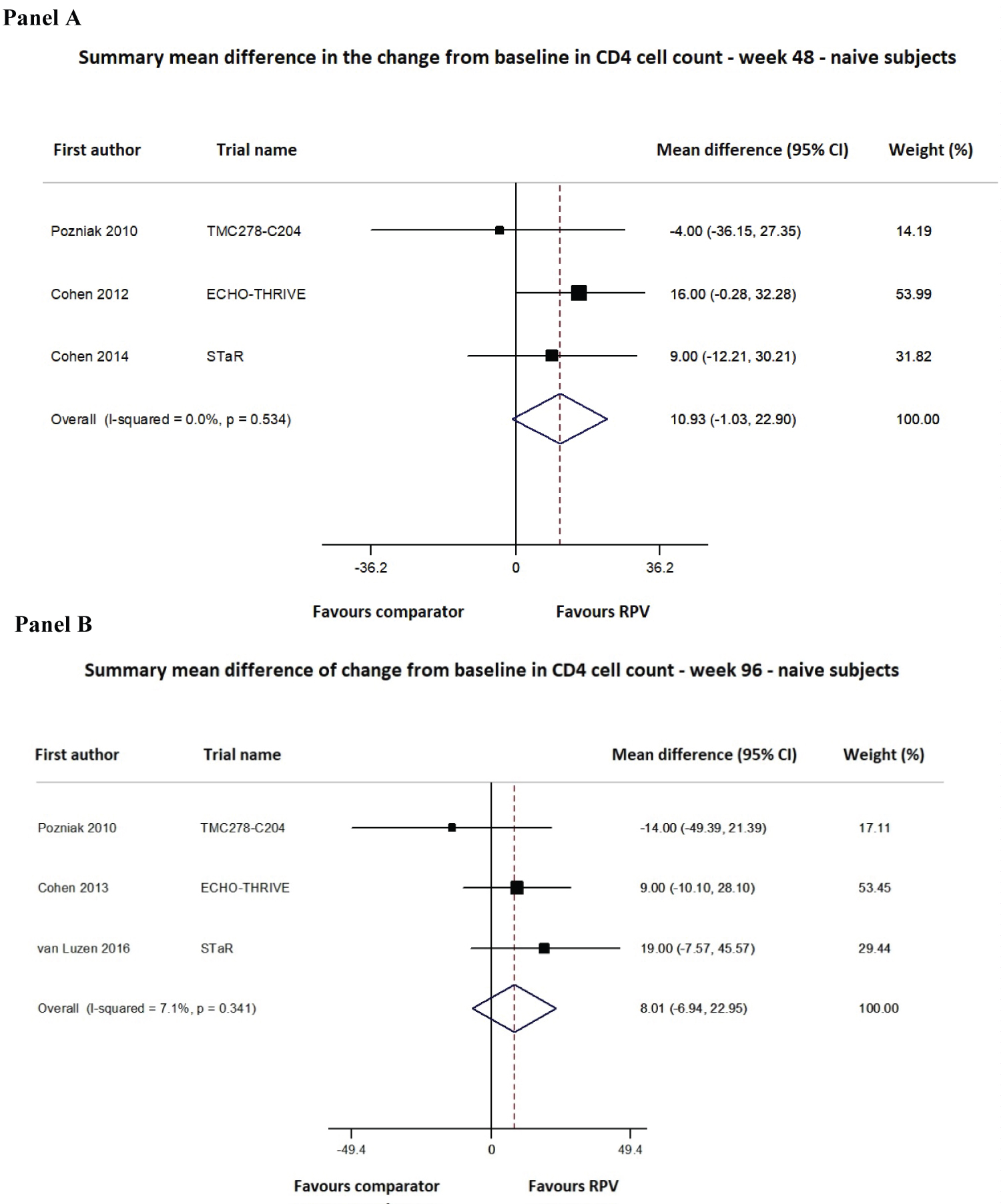 Figure 4: Meta-analysis of the change in CD4 cell counts from baseline for ART-naïve adult subjects at 48 (Panel A) and 96 (Panel B) weeks of follow-up.
View Figure 4
Figure 4: Meta-analysis of the change in CD4 cell counts from baseline for ART-naïve adult subjects at 48 (Panel A) and 96 (Panel B) weeks of follow-up.
View Figure 4
For cART-experienced virologically controlled patients, the mean and standard deviation at week 48 were available from 3 studies (1359 adult PLWH evaluated; Supplementary Table 1). Even in this group, the MD in the change in CD4 cell counts from baseline to week 48 did not differ between RPV and the comparator group (MD 14.01; 95% CI -1.62-29.64; Figure 5), and significant heterogeneity among the studies was not observed (Figure 5).
 Figure 5: Meta-analysis of the change in CD4 cell count from baseline for ART-experienced virologically controlled adult subjects at 48 weeks of follow-up.
View Figure 5
Figure 5: Meta-analysis of the change in CD4 cell count from baseline for ART-experienced virologically controlled adult subjects at 48 weeks of follow-up.
View Figure 5
Safety was primarily evaluated as the number of any adverse events (AEs), serious AEs, drug-related AEs and AEs leading to study discontinuation, using data at the longest follow-up and pooling together cART-naïve and cART-experienced patients; data were available from 8 (4549 subjects), 10 (4791 subjects), 9 (4515 subjects) and 12 studies (5467 subjects), respectively. As shown in Figure 6, Figure 7 and Figure 8 and Supplementary Figure 3, the RPV safety profile was not significantly different from that of the comparator according to any of the four assessed parameters. The pooled 48/96-week follow-up RRs for any AEs, serious AEs, drug-related AEs and AEs leading to study discontinuation were 1.01 (95% CI 0.97-1.05; Supplementary Figure 3), 1.04 (95% CI 0.78-1.39; Figure 6), 1.00 (95% CI 0.66-1.52; Figure 7) and 0.78 (95% CI 0.43-1.43; Figure 8), respectively. A moderate-high degree of heterogeneity was found among the studies’ RRs, except for the meta-analysis of any serious AEs (I2 48.7%, p = 0.069): any AEs (I2 66.8%, p = 0.010), drug-related AEs (I2 90.1%, p < 0.001) and AEs leading to discontinuation (I2 75.2%, p < 0.001; Figure 6, Figure 7 and Figure 8 and Supplementary Figure 3). No evidence of publication bias was detected (Supplementary Figure 2, panel B).
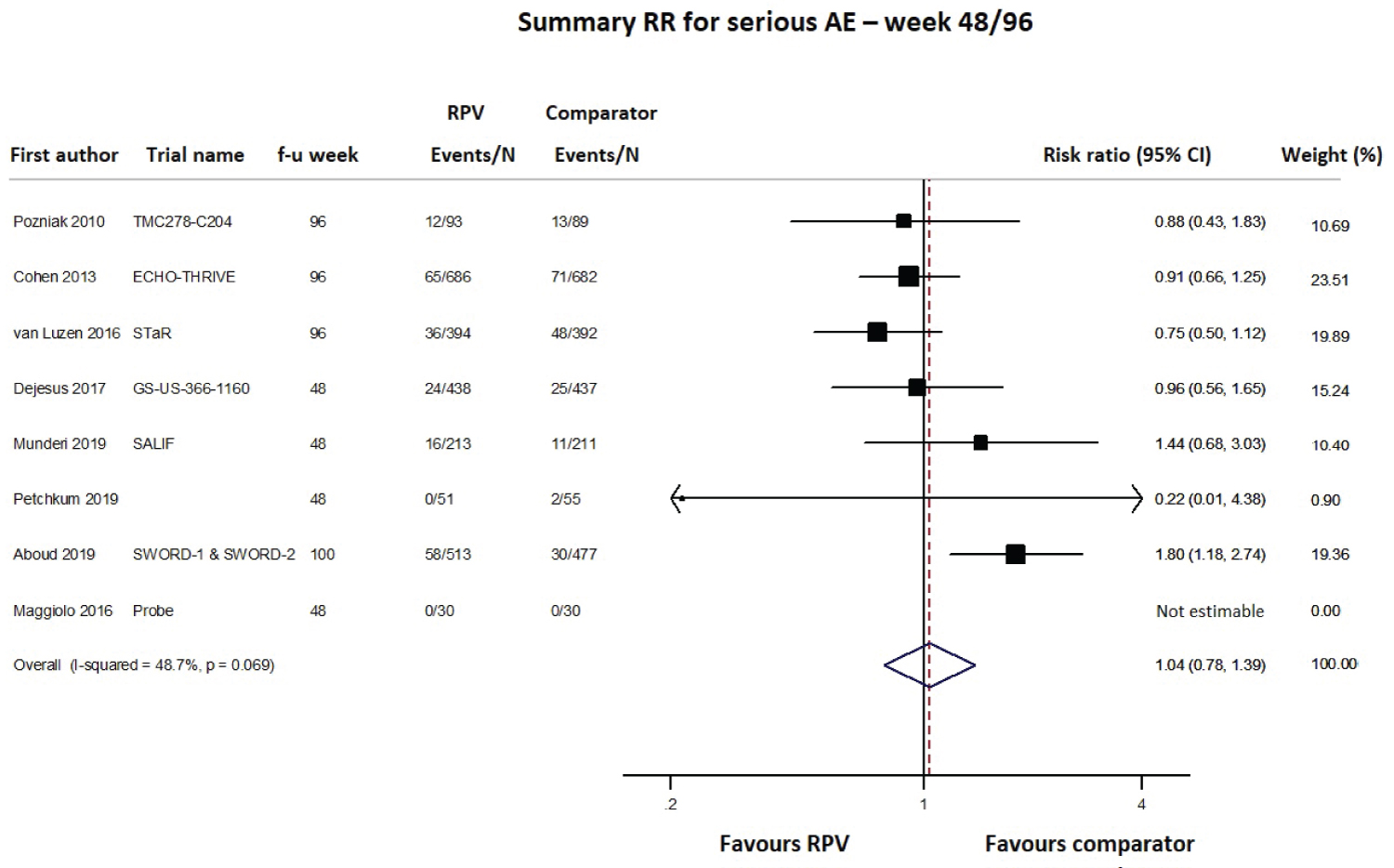 Figure 6: Meta-analysis of studies reporting data on any serious adverse events (AEs).
Figure 6: Meta-analysis of studies reporting data on any serious adverse events (AEs).
Note: Comparator group in the SWORD-1 & SWORD-2 trials: 52 weeks of ongoing ART followed by RPV/DTG (~48 weeks).
f-u: Follow-up.
View Figure 6
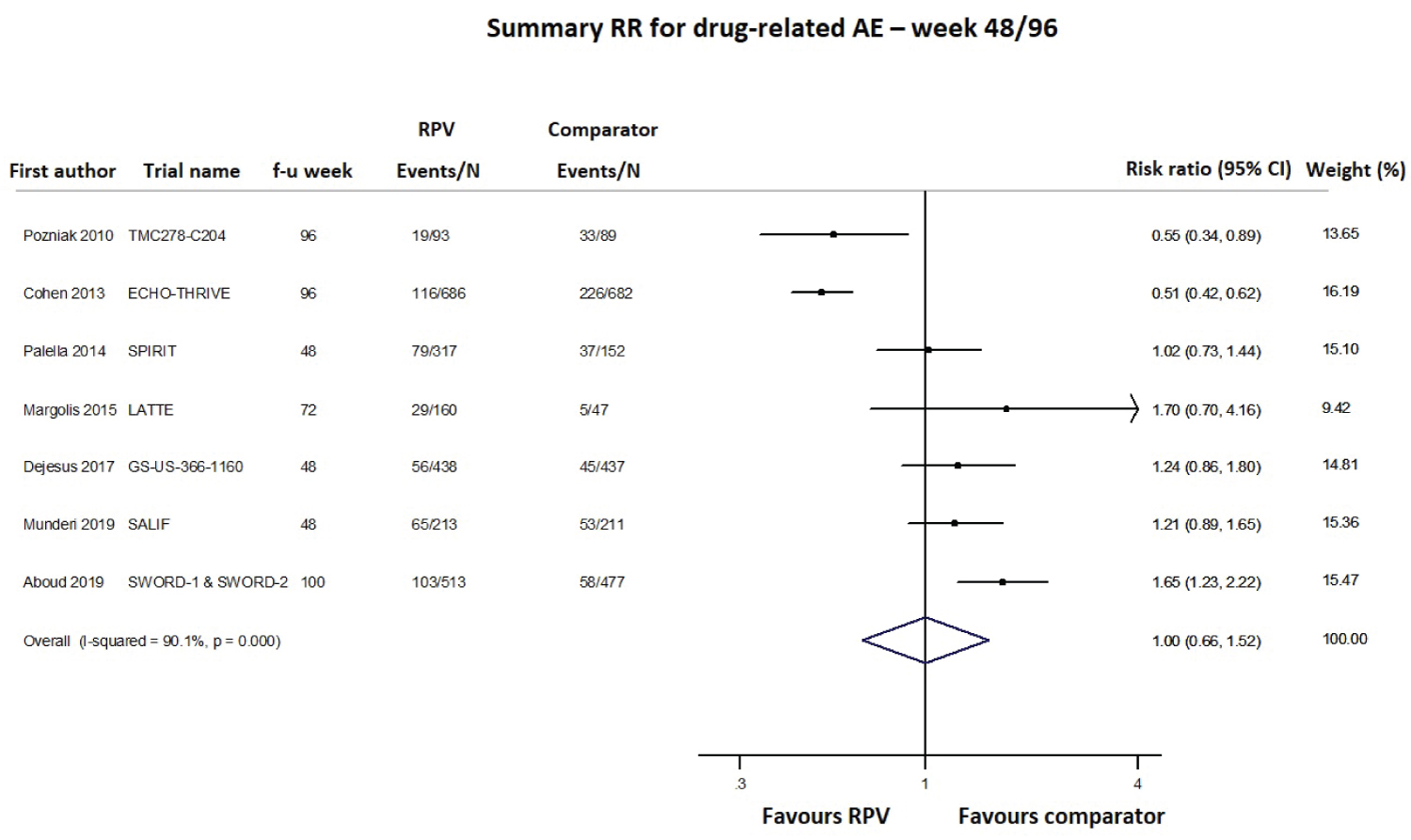 Figure 7: Meta-analysis of studies reporting data on drug-related adverse events (AEs).
Figure 7: Meta-analysis of studies reporting data on drug-related adverse events (AEs).
f-u: Follow-up.
Note: Comparator group in the SWORD-1 & SWORD-2 trials: 52 weeks of ongoing ART followed by RPV/DTG (~48 weeks).
Comparator group in the SPIRIT trial: 24 weeks of ongoing ART + 24 weeks of RPV/FTC/TDF.
View Figure 7
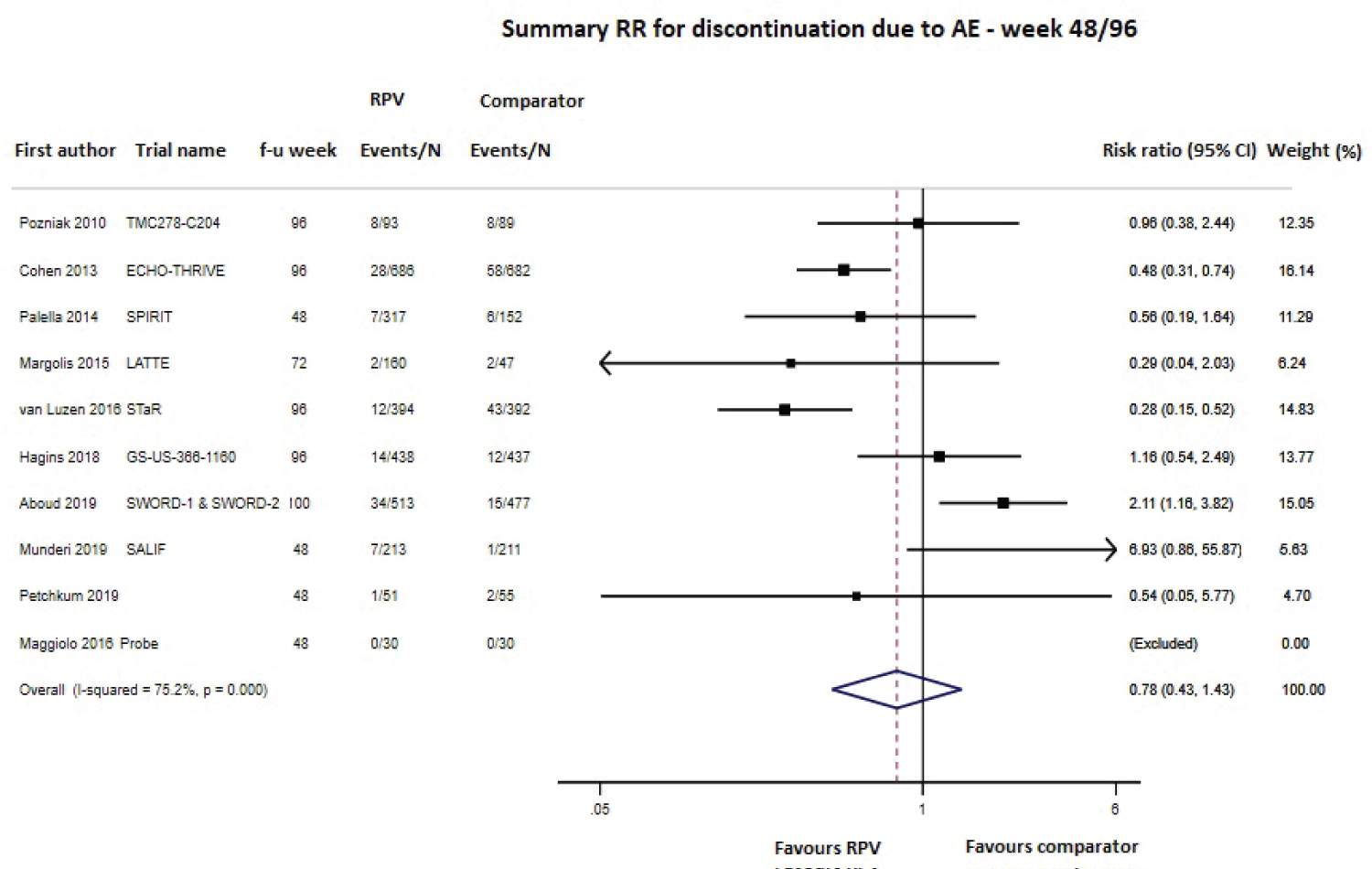 Figure 8: Meta-analysis of studies reporting data on discontinuation due to adverse events (AEs).
Figure 8: Meta-analysis of studies reporting data on discontinuation due to adverse events (AEs).
f-u: Follow-up.
Note: Comparator group in the SWORD-1 & SWORD-2 trials: 52 weeks of ongoing ART followed by RPV/DTG (~48 weeks).
Comparator group in the SPIRIT trial: 24 weeks of ongoing ART + 24 weeks of RPV/FTC/TDF.
View Figure 8
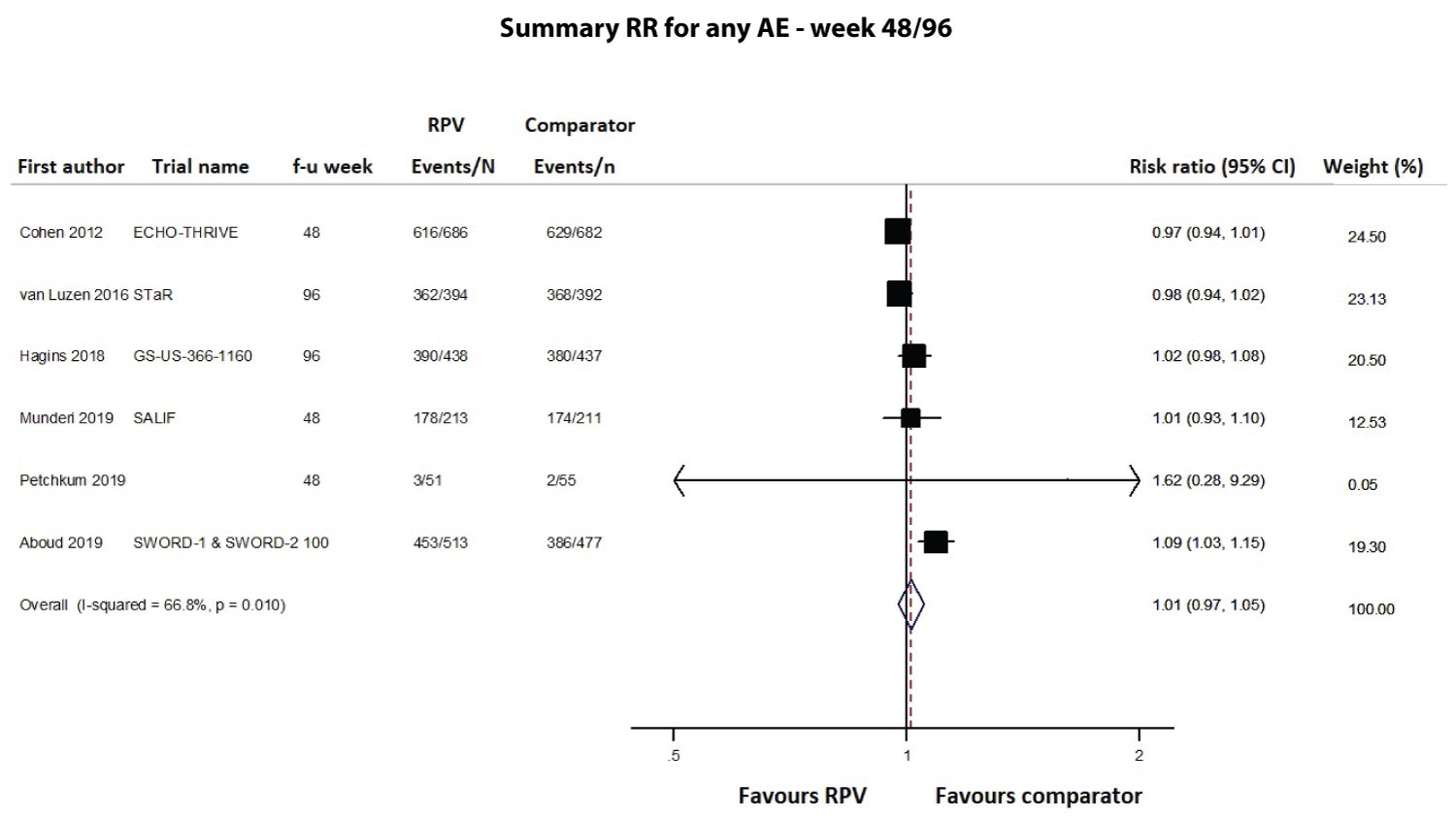 Supplementary Figure 3: Meta-analysis of studies reporting data on any adverse events (AEs).
f-u: Follow-up.
Supplementary Figure 3: Meta-analysis of studies reporting data on any adverse events (AEs).
f-u: Follow-up.
Note: Comparator group in the SWORD-1 & SWORD-2 trials: 52 weeks of ongoing ART followed by RPV/DTG (~48 weeks).
View Supplementary Figure 3
Available data from the included studies were not sufficient to perform a meta-analysis on the changes in lipid levels from baseline; therefore, we performed a systematic review on the changes in lipid levels from baseline at 48 weeks and at the longest follow-up (96 ± 4 weeks) for the cART-naïve and cART-experienced virologically controlled adult groups (Supplementary Table 2). For the former group, 3 (2154 subjects) [3,4,32] and 4 studies (1658 subjects) [5,7,33] were included for weeks 48 and 96, respectively. At week 48, total and LDL cholesterol showed no substantial variation in the RPV arms (from +1.0 to +3.09 and from -1.54 to +1.0 mg/dL, respectively), while there was a mild-to-moderate increase among the comparators (from +22.0 to +30.5 mg/dL, respectively). HDL cholesterol increased in both arms but more significantly in the comparator arms (from +2.0 to +4.25 mg/dL in RPV and from +8.0 to +10.4 mg/dL among the comparators). Triglycerides showed a mild reduction in patients treated with RPV (from -6.2 to -8.86 mg/dL) and an increase in patients treated with the comparator drugs (from +8.0 to +14.2 mg/dL). For all the studies, the changes in all these lipids from baseline differed between the arms at a statistically significant level (Supplementary Table 2).
At week 96, LDL changes spanned from +1.16 to +5.0 mg/dL in the RPV arms and from -5.0 to +29.0 mg/dL in the comparator arms; HDL changes varied from +2.0 to +6.0 mg/dL in the RPV arms and from +9.0 to +11.2 mg/dL in the comparator arms; and total cholesterol changes ranged from +3.0 to +10.0 mg/dL in the RPV arms and from +25.0 to +34.0 mg/dL in the comparator arms. For triglycerides, 3 studies [5,33] observed a reduction among patients in the RPV arms (from -6.20 to -8.0 mg/dL) and a mild-to-moderate increase among patients in the comparator arms (from +12.4 to +29.0 mg/dL), while the STaR study [7] failed to detect any differences in triglyceride changes from baseline between the RPV and comparator arms.
Six (2541 subjects) [9-11,14,34] and 3 studies (1865 subjects) [8,12] on cART-experienced virologically controlled subjects were included for weeks 48 and 96 (± 4 weeks), respectively. At week 48, all the lipids varied compared to their baseline levels, with a wide range among the studies. Specifically, LDL changes varied from -16.0 to +1.8 mg/dL in the RPV arms and from -14.0 to +8.52 mg/dL in the comparator arms. HDL changes spanned from -4.0 to +2.0 mg/dL in the RPV arms and from -4.0 to +5.16 mg/dL in the comparator arms, while total cholesterol changes ranged from -24.0 to +17.0 mg/dL in the RPV arms and from -24.0 to +7.25 mg/dL in the comparator arms. Triglyceride changes compared to baseline spanned from -64.0 to -4.0 mg/dL in the RPV arms and from -80.0 to +15.0 mg/dL in the comparator arms.
At the longest follow-up, LDL changes varied from -2.0 to +5.8 mg/dL in the RPV arms and from 0 to +6.18 mg/dL in the comparator arms. HDL changes spanned from -4.0 to +0.39 mg/dL in the RPV arms and from -1.16 to 0 mg/dL in the comparator arms, while total cholesterol changes ranged from -13.0 to +3.87 mg/dL in the RPV arms and from -3.0 to +2.32 mg/dL in the comparator arms. Data about changes in triglycerides were only reported by Hagins, et al. [12], which confirmed the significant difference favoring the RPV arm in total cholesterol reduction, already observed at week 48 [34] (Supplementary Table 2).
Twelve RCTs were included in this meta-analysis. The first RCTs started enrollment in 2005 and described cART-naïve subjects [6,33], while the most recent data from ongoing trials were published in 2019 and involved cART-experienced virologically controlled subjects [8]. In the ITT analysis of both the cART-naïve and experienced virologically controlled subjects, the virological response rate and the MD in the change in CD4 cell counts from baseline did not differ between the RPV and comparator arms, at either 48 or 96 weeks, with low variability among the studies. However, according to the baseline characteristics of naïve patients, compared to EFV, the difference in virological suppression favored RPV at both 48 and 96 weeks (+7% and +8%, respectively) among those starting cART with plasma HIV RNA < 100,000 cp/mL. Furthermore, among naïve patients starting cART with a CD4 count above 200 cells/μL, EFV reached a virological control rate similar to that of RPV only after 2 years of treatment. These results were also achieved in clinical practice in both naïve and cART-experienced patients with approximately 2 years of follow-up [15,16,19,21]. These results confirm previous evidence of RPV potency and efficacy in inhibiting a broad spectrum of HIV-1 genotypes and circulating recombinant forms, including K101E-, Y181C-, G190A- and K103N-mutated viruses, and in possessing generally lower half-maximal effective concentration values than those of other NNRTIs against specific HIV-1 isolates [43,44]. Notably, in 4 of the included studies, RPV was used in dual regimens (with dolutegravir, cabotegravir or darunavir/ritonavir) [8,9,11,28], and in 1 study, RPV doses were differentiated (either 25, 75 or 150 mg/day) [6,33]. In both cases, RPV potency in reaching viral undetectability was confirmed, as it was safe with no evidence of a dose-response relationship [33].
The safety profile of RPV was also similar to that of the comparator irrespective of the drug used (EFV, nevirapine or boosted protease inhibitors) in terms of both AEs and changes from baseline lipid levels. AEs leading to RPV discontinuation were relatively rare. In this analysis, pooling the naïve and experienced subjects could have introduced bias and increased the heterogeneity; the latter was also explained by the several different companions in RPV-anchored dual and three-drug regimens. In a previous meta-analysis, RPV showed lower rates of rashes and neurological AEs than EFV [30]; we have not performed subanalyses stratified according to the type of AEs; overall RPV has shown non-inferiority versus any control arm in the rate of drug-related AEs.
Among naïve patients at the longest follow-up, RPV was shown not to affect total and LDL cholesterol levels compared to the mild-to-moderate increase associated with the comparators and, despite its milder effect on a favorable increase in HDL cholesterol, the lowering effect on triglycerides in the RPV-anchored arm was in the opposite direction of the changes induced by the comparator drugs. Among cART-experienced virologically controlled subjects, RPV use in the first year from the switch was associated with a reduction in LDL and triglycerides (from a mild reduction to a mild increase in the comparator arms) and variable trends in HDL and total cholesterol (similarly to the comparators). At the longest follow-up, these differences were attenuated, with a significant persistence of lowered levels of total cholesterol. The results of this qualitative analysis of RCTs are in line with other studies that reported a neutral or favorable metabolic impact of RPV on lipids [45,46]. Interestingly, a recent phase IV RCT observed that patients switching from EFV to RPV (remaining on an FTC/TDF backbone) showed a decrease in the global amount of storage lipids and an increase in lysophosphatidylcholines and total steroids, resulting in an increment of metabolites with anti-inflammatory properties and a reduction in the repository of specific lipotoxic lipids [47]. Further studies are warranted to clarify the observed differential impact of RPV on lipid levels between cART-naïve and cART-experienced patients as well as the positive long-term consequences of its favorable metabolic impact on the several HIV-related comorbidities associated with alterations in lipid profiles, such as weight gain and HIV-associated neurocognitive disorders.
The main limitations of this meta-analysis are represented by: the variety of different comparators among the studies in cART-experienced patients; the patient data aggregation; the partially different time points for the efficacy/safety assessments; the limited data on lipid changes that did not allow us to perform a quantitative synthesis; the wide time-span of the included studies (2005-2019); the exclusion of non-English-language publications. Eight of the included RCTs were open-label, and thus, considered at an increased risk of performance bias. Nevertheless, since the assessed outcomes were objective measures, the risk of performance bias may still be restrained. Furthermore, our meta-analysis included only 4 studies using RPV-including dual regimens; only 1 out of the 8 studies assessing RPV-including regimens with tenofovir involved tenofovir alafenamide instead of disoproxil fumarate, and none reported data on long-acting injectable RPV. However, regarding the latter, two recent registrative studies conducted in naïve (after oral induction with dolutegravir and RPV) [48] and in virologically controlled subjects [49] have been recently published, showing the noninferiority of monthly injections of RPV plus cabotegravir to standard oral cART. Further analyses taking into account all these variables are warranted.
The main strength of our meta-analysis is the extensive retrieval of clinical studies from multiple databases based on a cross-checking strategy that allowed us to include additional potentially missed articles. Indeed, we updated a previous meta-analysis on 4 RCTs with 48 weeks of follow-up, [30] including data up to 96 (± 8) weeks and comparators other than only EFV. The adoption of the revised Cochrane risk-of-bias tool for randomized trials [37] improved the efficiency of previous tools in assessing the risk of bias, especially those arising from randomization, deviations from the intended interventions or outcomes measurement. The meta-analytic approach provides precise estimates of pooled results, which can inform and assist clinicians in routine practice. Furthermore, despite the studies time-span, patients characteristics did not significantly vary among the studies, the results following RPV treatments were consistent, and the heterogeneity was extremely low for both the efficacy outcomes. Lastly, the risk of publication bias was assessed and proven to be low, strengthening the reliability of our pooled effect measures.
This meta-analysis substantiates that RPV is an effective regimen for both cART-naïve and cART-experienced virologically controlled patients, observing no difference from the comparator arms. RPV proved to be also safe and well tolerated in both patient groups, with a similarly low rate of AEs and an advantageous impact on the lipid profile compared to the other treatment alternatives.
This work was supported by Janssen-Cilag SpA, Medical Affairs Department, Infectious Diseases; thanks to the collaboration of Mattia Trunfio (Research Fellow at the Unit of Infectious Diseases, Amedeo di Savoia Hospital, Department of Medical Science, University of Torino) in the drafting of the manuscript.
All the Authors contributed equally to this work and participated in manuscript preparation and revision.
Andrea Antinori (A.A.) has received honoraria for consultancies with Gilead Sciences, ViiV Healthcare, Merck Sharp & Dohme, Janssen-Cilag, Abbvie, and Bristol-Myers Squibb and has also received research grants from Gilead Sciences, Bristol-Myers Squibb, Janssen-Cilag, and ViiV Healthcare;
Adriano Lazzarin (A.L.) has received fees for advisory board participation and conference talks from BMS, ViiV, Gilead, MSD, Mylan, Abbvie, Janssen Cilag, and Teva;
Stefano Rusconi (S.R.) has received consulting fees and fee for advisory board participation from BMS, Gilead, MSD, ViiV, Janssen Cilag, Mylan;
Alessia Uglietti (A.U.), Daniela Mancusi (D.M.) and Roberta Termini (R.T.) are employees of Janssen-Cilag SpA, Italy;
Carlotta Galeone (C. G.) is consultant for Statinfo.