Proper statistical analysis is the most important thing in clinical trials if a person wants to come to accurate conclusions and make smart decisions about the safety and effectiveness of new medical interventions. The utilization of the Study Data Tabulation Model (SDTM) and the Analysis Dataset Model (ADaM) is imperative in facilitating this process. The Study Data Tabulation Model (SDTM) is a universally accepted and standardized framework utilized to structure and display data obtained from clinical trials. The utilization of a consistent structure for data representation facilitates the seamless integration and analysis of data derived from various studies. The Study Data Tabulation Model (SDTM) categorizes data into various domains, including but not limited to demographics, adverse events, and laboratory measurements. Variables within each domain are defined and coded using specific controlled terminology, ensuring consistency across different studies. The implementation of a standardized data structure facilitates the accessibility, comprehension, and analysis of data for statisticians, thereby mitigating the potential for errors and augmenting the overall quality of the statistical analysis. In contrast, the Analysis Dataset Model (ADaM) serves as a complementary framework to SDTM, with its primary objective being the preparation of datasets specifically tailored for statistical analysis. The main focus of the study is to examine statistical Analysis in Clinical Trials Using the Study Data Tabulation Model (SDTM) and the Analysis Dataset Model (ADaM). In addition, the study also efficiency and Time-Saving and impact on Data Quality.
Clinical trials, Study Data Tabulation Model (SDTM); Analysis Dataset Model (ADaM), CRF and FDA
New chemical entities and new drug formulations are tested in humans after completing laboratory trials in Phase I-IV studies to determine their safety and efficacy before receiving market approval. The procedures for conducting studies differ across protocols, with prequalified principal investigators overseeing the trials at research centres and gathering necessary data on source documents during multiple visits by the protocol [1]. The dataset encompasses various parameters such as subject demographics, habits, medications, events, study procedures, and test drug usage. The aforementioned information is commonly referred to as source data and constitutes an integral component of hospital records [2]. The investigators in charge of the study document the necessary trial information onto the Case Report Form (CRF), which may be in electronic or paper form, as provided by the sponsor or Clinical Research Organizations (CROs) [3]. The clinical data manager collaborates with the statistician, clinical operations team, medical monitor, and sponsor to develop the CRF [4]. To examine the ADaM (Analysis Dataset Model) it was found that the analysis data model specifies principles for the analysis data set and standards for a subject-level analysis file and for a basic data structure that can be used for a wild variety of analysis methods. This ADaM theory and application course provides an overview of the current implementation guide data structure and related material for submission. To focus on the course agenda mainly include the ADaM subject level analysis data set, ADaM basic data structure, ADaM occurrence data structure, ADaM implementation guide, ADaM supplemental document, and ADaM metadata that include time-to-event statistical examples and validation check. Lastly, the next course mainly includes additional material for submission the ADaM framework establishes standards for datasets and metadata, which facilitate the efficient production, duplication, and evaluation of statistical analyses in clinical trials. Additionally, it ensures a clear connection between analysis results, analysis data, and data represented in the Study Data Tabulation Model (SDTM), allowing for traceability. The ADaM standard is a mandatory requirement for the submission of data to regulatory authorities such as the FDA in the United States and the PMDA in Japan. The specific requirements for the Food and Drug Administration (FDA) can be found in the FDA's Data Standards Catalogue, which outlines the necessary guidelines for New Drug Applications (NDA), Abbreviated New Drug Applications (ANDA), and certain Biologics Licence Applications (BLA) submissions.
The development of an Analysis Data Model Implementation Guide (ADaM-IG) is undertaken concerning a particular ADaM model. Furthermore, there have been advancements in the development of supplementary ADaM materials that incorporate normative content tailored to specific analysis use cases. The present study focuses on a comprehensive list of the documents that were accessible during the release of ADaM-IG v1.3. It also provides an overview of how these documents are relevant to ADaM-IG Versions 1.0, 1.1, 1.2, and 1.3. In conjunction with Models and Implementation Guides, Conformance Rules have been devised to facilitate the adherence of generated data structures to established standards. The objective of these rules is to systematically identify and categorize all conformance rules and case logic found in ADaM documents. This process involves organizing and documenting these rules in a manner that facilitates quality processes and the development of tools.
To make focus on ADaM-IG v1.3 Release Package it describes all related documents that can be used with ADaM-IG v1.3 but it is not applicable. Secondly, ADaM-IG v1.2 Release Package describes all related documents that can be used with ADaM-IG v1.2 but it is not applicable as of now. In addition to this ADaM-IG v1.1 Release Package , it includes all related documents that can be used with ADaM-IG v1.1 and it is not applicable as of now. Lastly, ADaM-IG v1.0 Release Package Contains all related documents that can be used with ADaM-IG v1.0 and it is not applicable as of now. Additionally, the manager ensures that site personnel receive sufficient training to accurately complete the CRF by the established requirements [5]. During the development of the Case Report Form (CRF), the clinical data manager demonstrates a comprehensive understanding of the protocol and incorporates the CRF pages/forms through the scheduled visits, adhering to either sponsor standards or internal guidelines [6]. The Clinical Data Acquisition Standards Harmonization (CDASH) outlines standard procedures for the development of Case Report Forms (CRFs) that are widely accepted as being optimal, as opposed to utilizing individualized standards.
The primary objective of CDASH is to establish standardized content specifications for a fundamental collection of global Case Report Form (CRF) fields. The aforementioned CRF standards are global and apply to all therapeutic areas (TAs) throughout various phases [7]. The CDASH standard is a constituent of the Clinical Data Interchange Standards Consortium (CDISC) initiative. The work on CDASH standards was commenced by CDSIC in October 2006. The study groups were comprised of clinical data managers, statisticians, medical monitors, and programmers, who collaborated with organizations such as the Society of Clinical Data Management, National Cancer Institute, Association of CROs, FDA, CDISC, Critical Path Institute, and Cancer Biomedical Informatics Grid to gather data from clinical trial sites [8]. The data and recommendations were collected from the three International Conference on Harmonization regions, namely the USA, Europe, and Japan. The initial comprehensive version was made available for assessment in May of 2008. CDISC released the CDASH version 1.0 in October 2008, followed by the posting of version 1.1 in January 2011. The anticipated release date for Version 2.0 is slated for the year 2015.
The CDASH framework categorizes various data collection fields as highly recommended (HR) - The inclusion of a data collection field on the Case Report Form (CRF) is deemed necessary as per regulatory requirements [9]. The inclusion of specific data fields on a Case Report Form (CRF) is contingent upon certain conditions, such as the preference for a complete date of birth, which may not be feasible in certain regions. Additionally, the capture of adverse event (AE) time is only warranted if there exists another data point for comparison [10]. The utilization of the Study Data Tabulation Model (SDTM) has been deemed foundational in the development of the contents. CDASH 1.1's most recent iteration delineates the contents of 16 standard domains, as depicted in Figure 1.
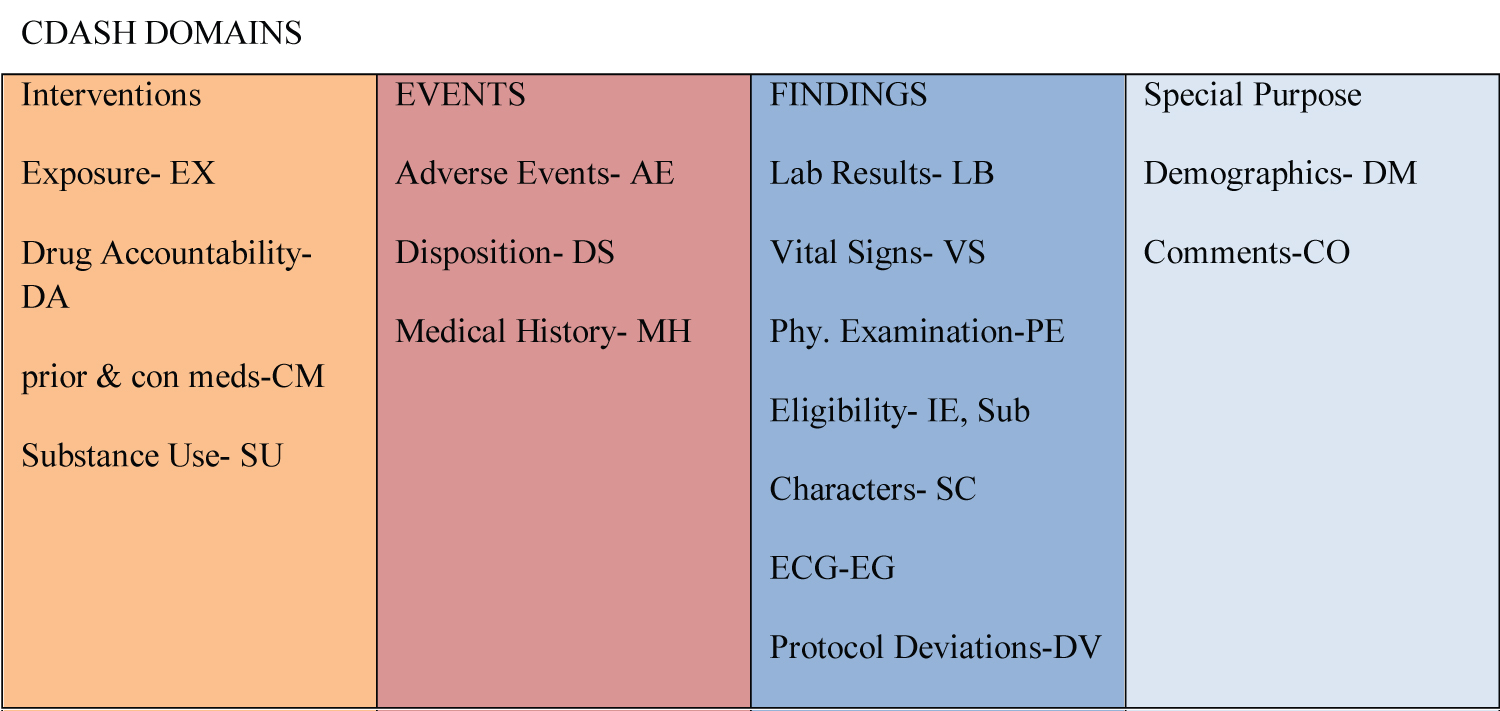 Figure 1: CDASH standard domains.
View Figure 1
Figure 1: CDASH standard domains.
View Figure 1
The CDASH protocol provides comprehensive information about the field name, prompt, field usage, guidelines for field completion, and recommended practices for each of the 16 domains (Table 1).
Table 1: CDASH domain tables structure. View Table 1
The meticulous construction of the database in the Study Data Tabulation Model (SDTM) is of utmost importance in the context of submitting data to regulatory authorities [11]. The utilization of raw data in the Study Data Tabulation Model (SDTM) format facilitates the generation of Common Technical Document (CTD) submissions and enables the creation of Analysis Dataset Model (ADaM) datasets with reduced programming requirements [12]. The CDASH standards, being a constituent part of SDTM, facilitate the seamless flow of data throughout the study by commencing data collection at the source, thereby eliminating any duplication of data collection [13]. It is highly recommended by CDASH to solely capture essential data that are necessary for statistical analysis. This methodology entails the removal of certain Conditional Random Field (CRF) categories, such as the compilation of binary responses for the Inclusion/Exclusion (IE) criteria CRF [14]. According to CDASH, the Inclusion and Exclusion criteria are applicable at the level of the study, and it is not mandatory to gather this data for every individual participant [15]. CDASH guidelines suggest that it is advisable to gather solely the unfulfilled criteria for which the deviation has received approval from the relevant parties (Figure 2).
 Figure 2: Traditional case report form for inclusion and exclusion criteria.
View Figure 2
Figure 2: Traditional case report form for inclusion and exclusion criteria.
View Figure 2
CDASH guidelines suggest that it is preferable to gather information on the status of physical examination tests rather than collecting data for individual body systems. Any anomalies that are detected should be documented in either the "medical history" or "adverse event" case report form [15]. The CDASH standards have been designed to prevent redundant data collection by eliminating certain types of CRFs, as illustrated in Figure 3.
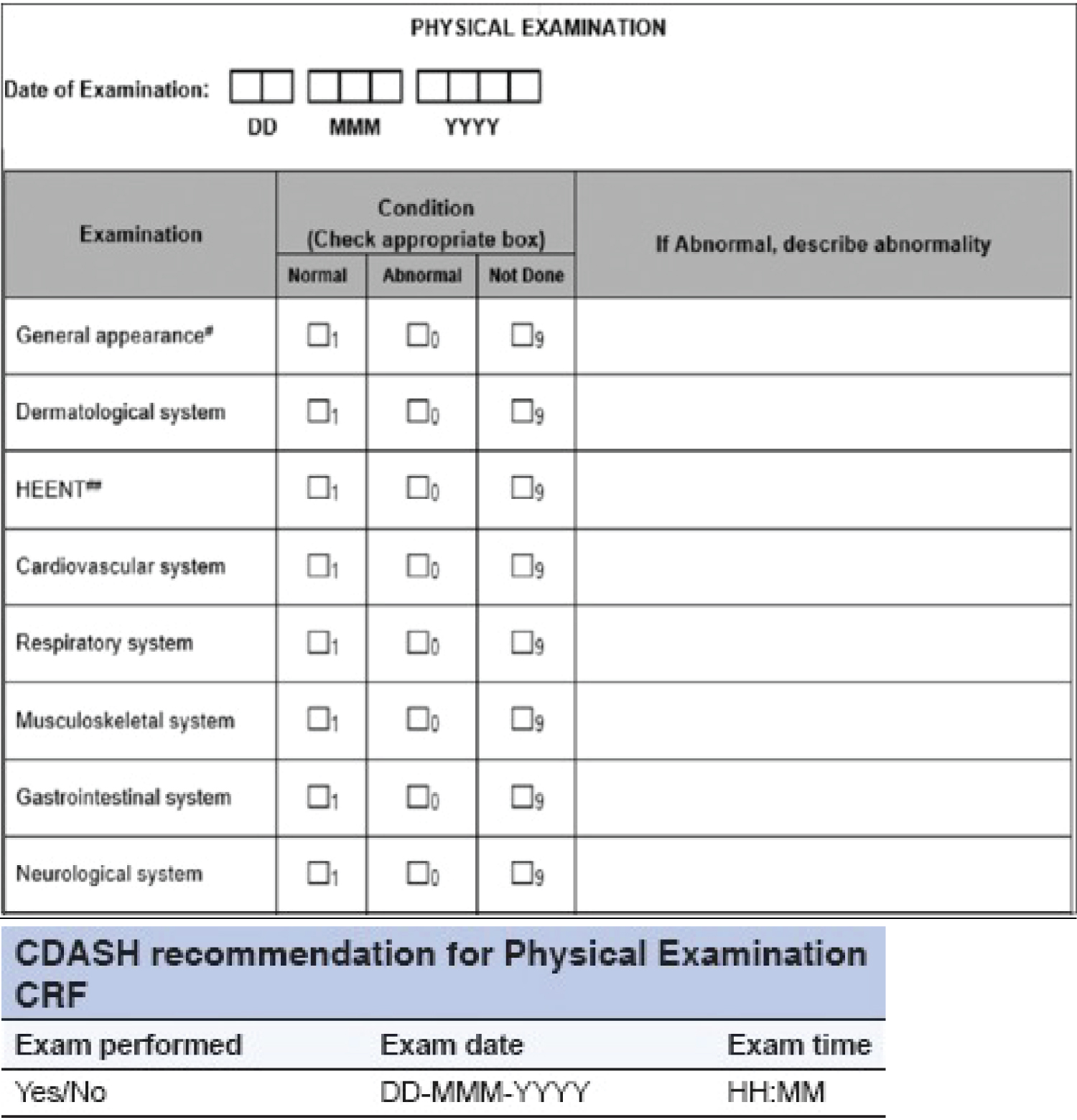 Figure 3: Traditional physical examination page.
View Figure 3
Figure 3: Traditional physical examination page.
View Figure 3
The Conditional Random Field (CRF) mentioned above eliminates the necessity of collecting and reconciling duplicated information by consolidating atypical data in a single centralized database. The decrease in the number of inquiries results in a reduction of the workload for both data administrators and on-site staff [12]. In addition, it enables the preservation of uniformity and standardization in the documentation of data for the designated objectives. Moreover, it reduces the need for coding, assuming that abnormalities in pulmonary embolism have already been coded [13] The adoption of CDASH Standards results in improved mutual understanding among the diverse stakeholders engaged in clinical trials, yielding higher quality data, reduced data queries, and simplified SDTM mapping for regulatory submissions.
The majority of Clinical Data Management System (CDMS)/Electronic Data Capture (EDC) tools, such as Medidata Rave, Oracle Clinical, Oracle Remote Data Capture, and CLIN Plus, enable the development of CDASH electronic Case Report Forms (CRFs) [16]. Tailored programming may be necessary for execution, especially when there is a necessity for amalgamation of laboratory data, electronic patient-reported outcomes, interactive web response systems, and electronic data capture [17]. The prerequisites are dependent on the complexity and approach of the investigation and the adherence to CDASH standards for continuous long-term trials may present a prospective difficulty [9]. The utilization of a customized approach is crucial to proficiently map out the various data points. The implementation of CDASH standards within a 5-year clinical trial that conforms to sponsor-specific protocols poses a significant challenge. The migration process necessitates a substantial allocation of resources towards the creation of documentation, mapping of fields to pre-existing fields, and other associated tasks. It is recommended to proactively determine the selection of data standards to be utilized [18]. The alignment of historical trial data with CDASH standards requires a customized approach.
The precise and focused development of user-friendly Case Report Forms (CRFs) accompanied by unambiguous instructions for their completion is a critical component of the initial stages of a research study protocol [19]. The preliminary Case Report Form (CRF) undergoes scrutiny by various stakeholders involved in the study, such as clinical research associates, data managers, medical monitors, and statisticians. The CRF that has been authorized serves as the basis for the programmers to set up the CRF within the EDC or CDMS tools that have been implemented for the study protocol [20]. Typically, the process of designing the case report form (CRF) and implementing the database for data entry requires a duration of 2-3 months. The CDASH domain Case Report Forms (CRFs) possess the potential for cross-study and cross-industry utilization without any restrictions on therapeutic areas (TAs). The creation of a repository of CDASH-defined CRFs can yield benefits for multiple studies, as it reduces the need for repeated efforts and saves time [21]. This, in turn, can lead to a reduction in operational costs associated with study start-up activities. The CDASH guidelines provide explicit specifications regarding the definitions of domains and their corresponding variables. The aforementioned elements consist of the variable name, definition, directives for clinical sites, execution, and fundamental configuration. The fundamental arrangement delineates the degree of necessity for the variable, ranging from highly recommended to recommended/conditional or optional. Nevertheless, CDASH does not delineate the length, sequence, and constituents of variables [22]. CDASH does not provide explicit definitions for code lists but instead relies on SDTM terminology. In cases where code lists are not included in the SDTM Implementation Guide, CDASH suggests the use of appropriate terms.
This study aims to examine the effects of the harmonization of clinical data acquisition standards on the utilization of full-time equivalents and associated costs. The task at hand involves the estimation and analysis of a given set of data. The finalization of CRFs and edit check specifications can be executed in compliance with the standards that are specific to the client, the Contract Research Organization (CRO) standards, which include Standard Operating Procedures (SOPs), or the Clinical Data Acquisition Standards Harmonization (CDASH) standards. The adoption of CDASH standards can lead to a reduction in the amount of time allocated to the development of case report forms, the specification of edit checks, the programming of screens, the implementation of edits, and the mapping of the Study Data Tabulation Model (SDTM) datasets [22]. The implementation of CDASH standards results in a reduction of review efforts by other study stakeholders concerning CRFs and completion guidelines [20]. A notable decrease of approximately 60% in full-time equivalent utilization is anticipated, resulting in a corresponding enhancement of operational efficiency and reduction of costs. The cost-effectiveness of implementing standards and the resulting savings, as well as the return on investment, are subject to variability based on a range of factors.
The clinical trial data submitted by pharmaceutical and biotechnology companies areanalyzed by regulatory body reviewers [22]. The review process for nonstandard data submission models typically spans a duration of around 18 months. The submission of data in CDSIC standardized formats is recommended by the FDA. The utilization of standard datasets enables regulators to expedite the review process by facilitating the reuse of their programs. Adherence to regulatory mandates necessitates the incorporation of CDISC standards from the outset, which confers significant advantages to pharmaceutical and biotechnology enterprises [23].
The CDISC standardization path:
CDASH → SDTM → ADaM → TLFs → CSR → eCTD (1)
The adoption of data standards, particularly CDISC standards, by the clinical research industry, can yield numerous advantages. It is recommended that technology firms offering data collection tools capitalize on the advantages of data standards and strive to optimize operational expenses [24]. Pharmaceutical companies allocate significant resources towards adhering to their internal protocols during the development of Case Report Forms (CRFs) and the creation of databases within electronic data capture (EDC) and clinical data management systems (CDMS). This methodology necessitates the utilization of data conversion techniques to comply with regulatory submission requirements [25]. It is advisable to pursue and execute the CDISC standardization pathway as a shared objective, as it offers advantages in terms of cost-effectiveness, time efficiency, and precision of data. Simultaneously, the implementation of CDASH standards serves as a facilitator in diminishing the duration of the review process, thereby enabling companies to introduce pharmaceutical products into the marketplace [23]. The establishment of standards is crucial for expediting the process of introducing orphan drugs and fast-track drugs to the market.
The SDTM Implementation Guide (SDTM-IG) for Human Clinical Trials is founded on the overarching framework of SDTM, which serves to structure clinical trial data intended for submission to regulatory agencies. The SDTM is constructed based on the fundamental notion of observations obtained from individuals who have taken part in a clinical investigation. Each data point can be characterized by a set of variables, which correspond to a particular row within a given dataset. Variables can be categorized based on their respective roles. The Role of a variable is responsible for specifying the nature of the information that it represents for individual observations and how it can be utilized. The utilization of identifier variables is crucial in academic research, as they serve to identify various aspects of the study, including the subject, domain, sequence number, and record. The topic variables are utilized to indicate the specific area of interest for the observation, such as the designation of a laboratory examination. Timing variables refer to the temporal aspects of observation, including the initiation and conclusion dates. Qualitative variables are characterized by descriptive text or numerical values that provide additional information about the observations, such as descriptive adjectives or units. The Trial Design model employs rule variables that specify an algorithm or executable method for defining the initiation, termination, branching, or looping conditions.
The qualifier variables can be classified into five distinct subclasses i.e., grouping qualifiers are employed to categorize a set of observations that fall under the same domain. Illustrative instances comprise --CAT and --SCAT. Result qualifiers refer to the specific outcomes that are linked to the variable of interest in a dataset of research findings and they respond to the inquiry prompted by the independent variable. The qualifying result indicators include --ORRES, --STRESC, and -STRESN. Synonym qualifiers are used to indicate an alternate nomenclature for a specific variable within an observation. The aforementioned instances comprise --MODIFY and --DECOD, which are synonymous with a --TRT or --TERM topic variable, and --TEST and --LOINC, which are interchangeable with a --TESTCD. The concept of Record Qualifiers pertains to the supplementary characteristics of an observation record in its entirety, as opposed to delineating a specific variable contained within the record. The aforementioned examples pertain to various SAE ag variables in the AE domain, as well as AGE, SEX, and RACE in the DM domain. Additionally, the Findings domain includes --BLFL, --POS, --LOC, --SPEC, and --NAM. Variable qualifiers are employed to provide additional modification or description of a particular variable within an observation. These qualifiers hold significance solely within the context of the variable they are associated with. The aforementioned instances comprise --ORRESU, --ORNRHI, and --ORNRLO, which are Variable Qualifiers of --ORRES. Additionally, --DOSU is a Variable Qualifier of --DOSE.
A domain is defined as a set of observations that are logically related and share a common topic and the rationale behind the association could be attributed to either the scientific content of the information or its function in the experiment [26]. A singular dataset is allocated to each respective domain and every domain dataset is characterized by a distinctive two-letter code that must be employed consistently during the submission process. The aforementioned code is contained within the SDTM variable designated as DOMAIN and serves four distinct purposes [4]. Firstly, it functions as the name of the dataset also it represents the value of the DOMAIN variable within the said dataset. Thirdly, it serves as a prefix for the majority of variable names in the dataset and finally, it is also utilized as a value in the DOMAIN variable [27]. Datasets are typically organized in a tabular format, with rows representing individual observations and columns representing different variables. The metadata definitions accompanying each dataset furnish details about the variables employed in the dataset. The metadata about the data is explicated in a data definition document, which is a Dene-XML document that is furnished to the regulatory authorities along with the data. The SDTM solely comprises the appellation, descriptor, and classification, accompanied by a concise set of CDISC directives that furnish a comprehensive overview of each variable [4]. The domain dataset models are incorporated in Section 5, which is dedicated to Models for Special Purpose Domains, and Section 6, which is focused on Domain Models. Please elaborate on the Controlled Terms or Format, provide guidelines on their appropriate usage, and furnish examples based on the General Observation Classes outlined in this document. Please refer to Section 1.4.1 titled "How to Read a Domain Specification".
It is recommended that the majority of subject-level observations gathered during the research be classified into one of the three general observation categories of SDTM, namely Interventions, Events, or Findings. The SDTM contains the lists of variables that are permissible to be utilized in each of these [28]. The Interventions course encompasses experimental, remedial, and alternative therapies that are given to the participant (with an actual or anticipated physiological impact) either as directed by the research plan (e.g., exposure to the investigational drug), concurrent with the study evaluation phase (e.g., accompanying medications), or self-administered by the participant (such as consumption of alcohol, tobacco, or caffeine). The Events category encompasses scheduled protocol milestones, such as randomization and study completion, as well as unscheduled events, conditions, or incidents that occur independently of planned study evaluations during the trial, such as adverse events, or before the trial, such as medical history [27]. The Findings category encompasses the data obtained from deliberate assessments aimed at addressing particular inquiries or tests, such as laboratory analyses, electrocardiogram evaluations, and queries listed on survey forms.
The SDTM comprises four distinct categories of datasets in addition to those that are founded on the overall observation classes. Domain datasets comprise subject-level information that does not adhere to any of the three overarching categories of observations [29]. The aforementioned categories, namely Demographics (DM), Comments (CO), Subject Elements (SE), and Subject Visits (SV), have been explicated in Section 5, which pertains to Models for Special Purpose Domains. The Trial Design Model (TDM) datasets pertain to the study design and do not comprise any subject data [30]. The aforementioned datasets, namely Trial Arms (TA) and Trial Elements (TE), are expounded upon in Section 7 of the Trial Design Model Datasets. The investigation of reference datasets is recommended, encompassing Device Identifiers (DI), Non-host Organism Identifiers (OI), and Pharmacogenomic/Genetic Biomarker Identifiers (PB) [31]. These structures offer a means of representing subject-specific terminology utilized in study data.
It is recommended that a sponsor exclusively provide domain datasets that have been gathered or derived directly from the gathered data for a particular study [26]. The selection of data to be gathered ought to be grounded on the scientific goals of the research, rather than being solely dictated by the SDTM. It should be noted that any data that is gathered and intended for submission in an analysis dataset must also be present in a tabulation dataset [32]. The data gathered for a particular research endeavor may employ conventional domains from this and other SDTM Implementation Guides, in addition to supplementary personalized domains derived from the three overarching categories of observations. Section 3.2.1, Dataset-Level Metadata, furnishes a roster of conventional domains [2]. The definitive domains will solely be made available through an SDTM Implementation Guide, which may include the SDTMIG for human clinical trials or an alternative implementation guide, such as the SDTMIG for Medical Devices.
The subsequent segment delineates the comprehensive procedure for generating a personalized domain, which necessitates adherence to one of the three general observation classes of SDTM. The number of domains presented ought to be contingent upon the particular prerequisites of the investigation [33]. The creation of a custom domain is contingent upon the distinctiveness of the data and its incompatibility with any pre-existing published domains. People should focus on the SDTM Draft Domains section on the CDISC wiki page titled "SDTM Draft Domains Home." Search for a pre-existing and pertinent domain model that can function as a prototype. If no pre-existing model appears suitable, one should opt for the general observation category (Interventions, Events, or Findings) that most accurately corresponds to the data by taking into account the subject matter of the observation [34]. The customary method for choosing variables for a personalized domain is outlined below (Figure 4).
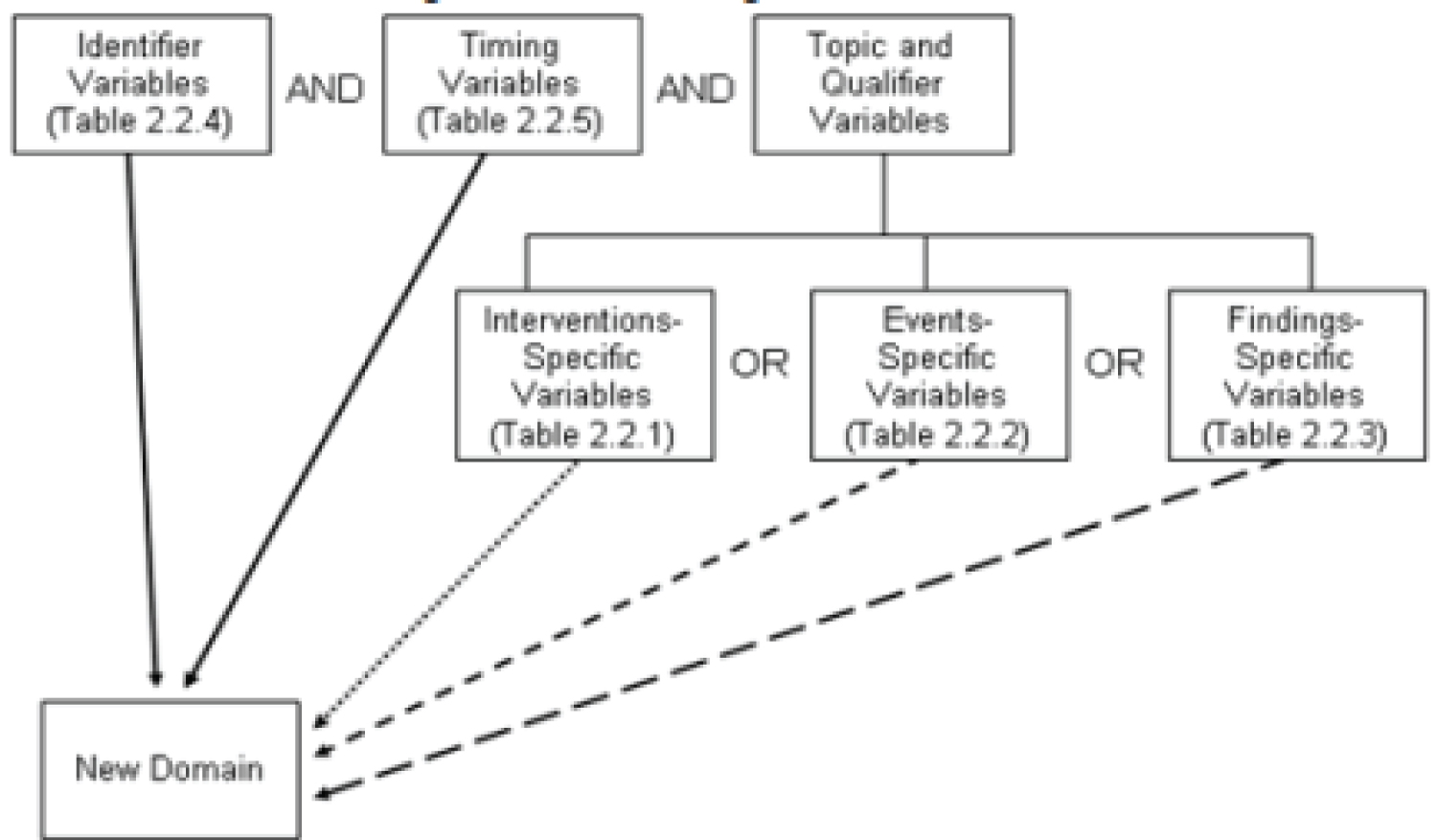 Figure 4: Creating a new domain.
View Figure 4
Figure 4: Creating a new domain.
View Figure 4
The formulation of analysis datasets is primarily influenced by the scientific and medical goals of the clinical trial. One of the core principles entails ensuring that the arrangement and substance of the analysis datasets facilitate the effective and unequivocal transmission of the scientific and statistical elements of the clinical trial [35]. The primary objective of ADaM is to establish a comprehensive framework that facilitates the analysis of data, while concurrently ensuring that reviewers and other recipients of the data possess a lucid comprehension of the data's lineage, spanning from its collection to the subsequent analysis and resulting outcomes [36-38]. The ADaM datasets serve as the primary and authoritative repository for all data derivations employed in statistical analyses. The utilization of standardized analysis datasets and metadata offers significant advantages to data recipients, extending beyond the realms of effective communication and transparency [39,40]. ADaM datasets integrate both derived and collected data from various SDTM domains, other ADaM datasets, or a combination of both, into a single dataset. This integration allows for analysis to be conducted with minimal or no additional programming required. To facilitate statistical analysis with minimal programming, analysis datasets must be accompanied by metadata and are prepared in an analysis-ready format, possessing both appropriate structure and content. Survival analysis is a commonly employed set of statistical techniques that are utilized to examine the incidence and temporal patterns of events [41-43]. The time to an event is often a focal point of interest in numerous clinical studies. Survival data possesses a distinctive characteristic wherein, upon the conclusion of the designated observation period, the event of interest may not have transpired for all individuals. Hence, the duration until the occurrence of an event for these individuals is considered as censored, specifically right-censored, and individuals may experience censoring due to diverse factors [44]. The ADaM standards propose the utilization of a collection of Time-to-Event Variables to capture the status of censoring about a specific event of interest, including the date of censoring and the underlying reason [45,46]. The structure of the ADaM TTE (time-to-event) analysis dataset is specifically designed to facilitate the application of widely used time-to-event analysis techniques, including the Kaplan-Meier Estimation and Cox Proportional Hazard Model.
The structure of this ADaM dataset bears similarity to the SDTM domain TR, as it follows a pattern of one observation for each subject, timepoint, tumor, response assessment criteria, and evaluator. Following the initial identification of baseline lesions, which include both Target and Non-Target lesions, during the screening process, any subsequent information regarding newly identified lesions is recorded and stored in the TU database [1,2,47]. The representation of this concept in ADTR is accomplished through the utilization of a dual set of records, one for documenting the date and time of the initial occurrence, and another for documenting the date and time of the definitive evaluation. Two parameters related to the New Lesion are recommended due to the potential variability between the timepoint of initial appearance and the timepoint of unequivocal assessment. The SDTM domain TR includes data about the sum of diameters of target lesions at each time point. It is recommended to store timepoint level information in the ADaM dataset called ADTRS.
The dataset provided encompasses results at the level of individual time points and each subject is observed once at each time point for each parameter, response assessment criteria, and evaluator. The inclusion of qualifiers, such as information about the Evaluator, is prohibited in the BDS structure [4,48]. Consequently, this information has been incorporated into the parameters, as exemplified by "Timepoint Overall Response by Independent Reviewer." The variable AVAL should be utilized for the first three parameters, as they yield a numeric result. The final four parameters are noteworthy in that the outcome is a character value. Therefore, it is advisable to employ the variable AVALC. The determination of whether specific observations are included or excluded from future analysis holds significant importance. The ADaM methodology employs analysis flags (ANLzzFL, where ZZ represents a two-digit numerical value, such as 01) sequentially to signify observations that meet specific criteria for one or more analyses [49,50].
Sensitivity analyses are frequently conducted to assess the impact of varying censoring rules on efficacy endpoints, which may differ from those employed in the primary analysis. In such instances, it is necessary to employ supplementary analysis indicators to identify and encompass the data entries that are encompassed within each sensitivity analysis. For instance, the code ANL02FL may be generated to facilitate sensitivity analysis. In this case, the calculation of the progression-free survival (PFS) is influenced by variations in the censoring rule, specifically regarding the inclusion of overall time-point evaluations that occur after multiple instances of missing adequate assessments. A supplementary analysis indicator denoted as ANL03FL, may be established to conduct a sensitivity analysis in which the selection of time points utilized in the derivation process is guided by the Immune-Related Response Evaluation Criteria. It is advisable to utilize an additional set of analysis indicators to detect the initial instance of a responder's response, whether it be a complete or partial response, denoted as 'CR' or 'PR' respectively [51]. Before assigning values to these flag variables, it is crucial to consult the SAP (System Applications and Products) to ascertain whether confirmation of response is necessary and what specific criteria must be met for confirmation. For instance, in situations where confirmation is necessary, consider a scenario involving a subject with a post-baseline sequence of timepoint overall responses denoted as {‘SD’, ‘PR’, ‘SD’, ‘SD’, ‘PR’, ‘PR’, …}, where ‘SD’ represents "Stable Disease". In this case, the fifth post-baseline evaluation should be identified with a flag, as it signifies the initial confirmed response. In the case of another subject with post-baseline timepoint overall responses categorized as 'SD', 'PR', and 'PD', no records will be annotated due to the absence of a confirmed 'PR' response. Consequently, this subject cannot be deemed a responder as per the Statistical Analysis Plan (SAP) [3]. In a manner akin to the aforementioned analysis flags, supplementary analysis flags may be generated, as necessary, for various sensitivity analyses. It is advisable to calculate an additional variable, denoted as PCHG (Percent Change from Baseline), for the parameter 'Timepoint Sum of all Target Lesion Diameters (mm)' at each timepoint following the baseline. The information could potentially be utilized in the Analysis Dataset for Efficacy (ADEF) to calculate the Percent Change in the Sum of Target Lesion Diameters from Baseline to Post-baseline Nadir (the lowest point). This data is commonly visualized in a widely employed waterfall plot.
The BDS dataset should include the derived efficacy parameters, where the presence of censoring does not affect the analysis. The dataset has been constructed with each observation representing a subject, subject-level parameter, response assessment criteria, and evaluator. The inclusion of qualifiers, such as information about the Evaluator, is not permitted in the BDS structure. Consequently, this information has been incorporated into the parameters, as exemplified by "Best Overall Response by Independent Reviewer." The ADaM dataset primarily includes two subject-level parameters: 'Best Overall Response' (BOR) and 'Percent Change in Sum of Target Lesion Diameters from Baseline to Post-baseline Nadir' (Yamamoto, et al. 2018). The initial parameter is calculated for all subjects included in the intent-to-treat analysis and the derivation of the second parameter is limited to subjects who have both baseline measurements and at least one post-baseline measurement of the sum of target lesions’ diameters in the ADTRS dataset, as indicated by the corresponding analysis flag. The values of these parameters will be stored respectively in variables AVAL or AVALC. The algorithm utilized for the derivation of the Best Overall Response is outlined in the SAP (Study Protocol). In certain instances, a research investigation may necessitate the computation of the benefit-to-cost ratio (BOR) both with and without confirmation of response, or the utilization of distinct evaluation criteria. In instances of this nature, it is advisable to allocate multiple parameters within this dataset. The determination of the value of BOR is a prerequisite for the calculation of ORR, which serves as a widely employed measure of efficacy. The use of previously established alternative disease-related treatment responses (ADTRS) is not suitable for replacing alternative disease evaluation forms (ADEF) in the derivation of baseline of response (BOR) [52]. This is due to several scenarios, such as instances of early stable disease (SD) where the subject only has an SD assessment after the baseline, which is considered too early to be counted as the BOR for SD. Additionally, the absence of post-baseline evaluations or the presence of only one post-baseline evaluation (PR) when confirmation of response is necessary also precludes the use of ADTRS. In instances of this nature, the value of BOR may deviate from the findings of any of the assessments conducted at different time points. Likewise, when multiple evaluation criteria are employed, it is anticipated that several parameters will be utilized for the "Percent Change in Sum of Target Lesion Diameters from Baseline to Post-baseline Nadir," with each parameter corresponding to a specific criterion.
Moreover, the inclusion of Disease Control Rate (DCR) is typically incorporated as an exploratory endpoint in the study. The Disease Control Rate (DCR) refers to the percentage of subjects within the Analysis Set who exhibit a Best Overall Response (BOR) of 'Complete Response' (CR), 'Partial Response' (PR), or 'Stable Disease (SD). To determine the values for Objective Response Flag (EFORFL) and Disease Control Flag (EFDCFL) for the parameter of 'Best Overall Response', it is recommended to include two additional custom flags. These flags, when populated, will facilitate the calculation of the Overall Response Rate (ORR) and Disease Control Rate (DCR) [53]. It is advisable to include an additional variable, BORDESC (Best Overall Response Description), which is exclusively populated for the parameter(s) Best Overall Response. This variable provides the reasoning behind the assignment of the Best Overall Response, with potential values such as 'Confirmed CR', 'Unconfirmed PR', 'Early SD', and so on.
The ADTTE dataset should include the efficacy parameters that necessitate censoring and the inclusion of these parameters represents a key attribute of clinical trials in the field of oncology. The dataset has been constructed to have one observation for each subject, time-to-event subject-level parameter, response assessment criteria, and evaluator. The inclusion of qualifiers, such as information about the Evaluator, is not permitted in the BDS structure. Consequently, this information has been incorporated into the parameters, as exemplified by "Progression Free Survival (months) by Investigator." As previously stated, the majority of efficacy endpoints in the field of oncology involve time-to-event analysis with censoring. For instance, three parameters that are frequently employed are "Overall Survival Time (Months)", "Progression Free Survival Time (Months)", and "Duration of Response (Months)". The initial two parameters are calculated for all subjects included in the intent-to-treat analysis, while the third parameter is only calculated for subjects who achieved a complete response (CR) or partial response (PR) in the ADaM dataset ADEF [54,55]. These subjects are identified by having a value of 'Y' in the EFORFL variable in ADEF. In certain research endeavors, it may be necessary to compute time-to-event parameters utilizing various assessment criteria or employing rules based on sensitivity analysis. In instances of this nature, it is advisable to allocate multiple distinct parameters within this dataset. The values of the parameters will be stored in variables AVAL and CNSR, by the CDISC Guidance "ADaM Basic Data Structure for Time-to-Event Analysis, Version 1.0." The ADTTE should include additional significant variables, namely ADT (Analysis Date), STARTDT (Time to Event Origin Date for Subject), EVNTDESC (Event or Censoring Description), and CNSDTDSC (Censor Date Description) [56]. The methodology for deriving all of these parameters should be outlined in the System Analysis and Programme Development (SAP) documentation. If the study endpoint includes the Clinical Benefit Rate (CBR), which represents the proportion of subjects in the Analysis Set who achieve a Best Overall Response (BOR) of 'Complete Response' (CR), 'Partial Response' (PR), or have durable stable disease, it is recommended to establish two custom flags: EFDUSDFL (Durable Stable Disease Flag) and EFCBFL (Clinical Benefit Flag). These flags will be used to assess the parameter of 'Progression Free Survival Time'. The designation of "Durable SD" is exclusively assigned to subjects with a Baseline of Response (BOR) categorized as 'SD' and with a minimum duration of SD that is equal to or greater than xx weeks. The specific value of xx should be determined by the protocol and Statistical Analysis Plan (SAP). Moreover, the duration of stable disease (SD) is a concept based on time-to-event, employing the identical censoring rule as progression-free survival (PFS) [57]. The duration of stable disease (SD) is equivalent to the length of time during which subjects with the best overall response (BOR) of SD do not experience disease progression. It should be noted that the creation of EFDUSDFL is limited to subjects with a BOR (basis of record) designation of 'SD'. Similarly, the marking of EFCBFL with 'Y' is contingent upon the subject having either a value of 'Y' for ADEF.EFORFL or a value of 'Y' for EFDUSDFL (Figure 5).
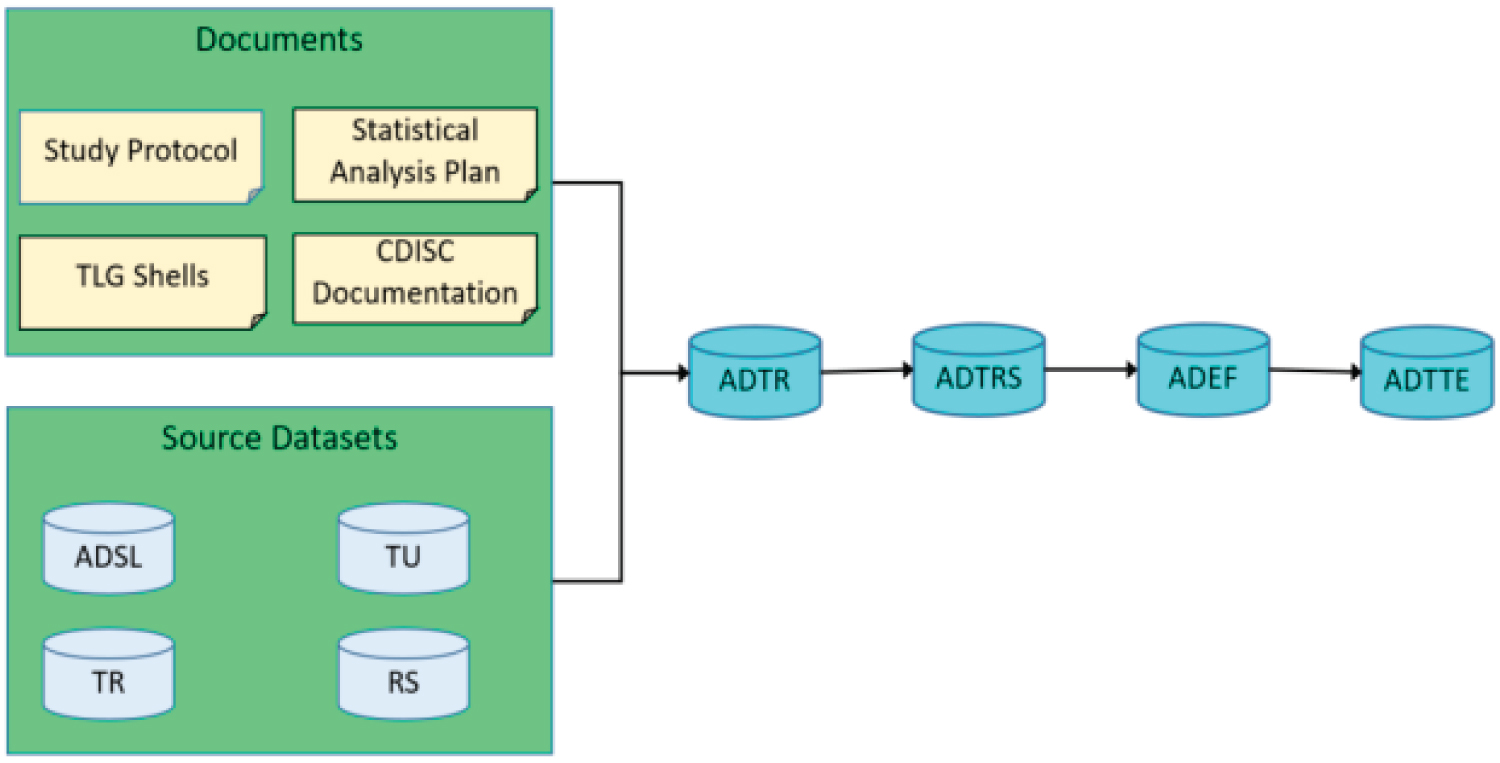 Figure 5: Source and order of generating efficacy ADaMs.
View Figure 5
Figure 5: Source and order of generating efficacy ADaMs.
View Figure 5
The utilization of the Study Data Tabulation Model (SDTM) and the Analysis Dataset Model (ADaM) in clinical trials holds significant importance in the assessment of novel medical interventions and treatments through statistical analysis. Standardized data models play a crucial role in maintaining consistency and accuracy within clinical trial data. By adhering to these models, researchers and regulatory authorities can effectively interpret and compare results [58]. The implementation of SDTM and ADaM has yielded various advantages for the clinical research community, encompassing enhanced data quality, heightened transparency, and improved traceability. Consequently, these outcomes contribute to the generation of more dependable and credible conclusions. The utilization of SDTM and ADaM has been found to have a significant impact on the facilitation of data integration and analysis across various studies and therapeutic domains [4]. These models offer a standardized framework for the representation of data, thereby enabling the aggregation and examination of data from various origins. Consequently, scholars can perform meta-analyses and cross-study comparisons more conveniently, thereby enhancing the breadth of knowledge and the applicability of research findings [5]. The implementation of this standardized approach has additionally facilitated the expeditiousness of the review process conducted by regulatory agencies, thereby simplifying the evaluation of the safety and effectiveness of novel medical interventions intended for patient utilization.
Nevertheless, the implementation of SDTM and ADaM in clinical trials is not devoid of challenges and the conversion of unprocessed data into Standard Data Tabulation Model (SDTM) and Analysis Data Model (ADaM) formats can prove to be a laborious and demanding task, particularly in the context of intricate and extensive clinical trials. Furthermore, the task of maintaining data consistency and accurately mapping variables to their respective domains can pose significant challenges, necessitating comprehensive quality checks, and validation processes. In addition, it is important to note that certain antiquated legacy systems and databases may not possess complete compatibility with the SDTM and ADaM standards. Consequently, data transformations and integration endeavors become necessary, which, if not executed with due care, can potentially give rise to errors [59]. To surmount these challenges, it is imperative to foster collaboration among various stakeholders, encompassing statisticians, data managers, and IT experts. Enhancing efficiency and accuracy in data conversion and analysis can be achieved through the investment in training and education for the team on the appropriate utilization of SDTM and ADaM. Automation tools and software solutions that are specifically developed for SDTM and ADaM can be beneficial in decreasing the amount of manual work required and ensuring compliance with established standards. By utilizing external expertise, such as engaging consultants who possess extensive experience in the implementation of the Study Data Tabulation Model (SDTM) and Analysis Data Model (ADaM), organizations can effectively address challenges and enhance their adherence to regulatory obligations [6,60]. The utilization of SDTM and ADaM in statistical analysis of clinical trials has notably enhanced the Caliber and uniformity of data across diverse studies, thereby resulting in more resilient deductions regarding the safety and effectiveness of medical interventions.
The present discourse acknowledges the prevalent utilization of the Study Data Tabulation Model (SDTM) and the Analysis Dataset Model (ADaM) within the realm of statistical analysis of clinical trials. However, it is imperative to highlight the existence of a discernible lacuna in the existing body of research, thereby necessitating further investigation and scholarly scrutiny. An area of utmost importance necessitating additional inquiry pertains to the enduring ramifications of employing the Study Data Tabulation Model (SDTM) and Analysis Data Model (ADaM) on the overall efficacy and cost-efficiency of clinical trial procedures. The recognition of the positive impact of standardized data models, such as SDTM and ADaM, on enhancing data quality and transparency is widely accepted. However, there remains a necessity for empirical investigations to evaluate the extent to which the implementation of these models affects various stages of the clinical trial lifecycle, encompassing data collection, analysis, and regulatory submissions [61]. Furthermore, the existing literature reveals a notable research gap in the assessment of the scalability and adaptability of the Study Data Tabulation Model (SDTM) and Analysis Data Model (ADaM) in various clinical trial settings and therapeutic domains.
The utilization of statistical analysis within the context of clinical trials holds significant importance in the assessment of the safety and effectiveness of novel treatments or interventions. The analysis of clinical trial data is facilitated by two prominent frameworks, namely the Study Data Tabulation Model (SDTM) and the Analysis Dataset Model (ADaM). These models offer standardized structures for the organization and analysis of data, guaranteeing uniformity and the ability to make comparisons across various studies. The utilization of the Study Data Tabulation Model (SDTM) in clinical trials serves the purpose of ensuring the harmonization and standardization of data obtained from diverse sources, thereby facilitating its transformation into a uniform format [62]. The Study Data Tabulation Model (SDTM) offers a standardized framework for the organization and categorization of data variables, facilitating the seamless integration and dissemination of data. This approach not only facilitates the simplification of the analysis procedure but also allows for the efficient aggregation of data and the conduct of meta-analyses across numerous studies. By adhering to the principles of the Study Data Tabulation Model (SDTM), various researchers can effectively interpret and validate the outcomes of their analyses, thereby improving the dependability and replicability of their findings [63]. Additionally, the ADaM (Analysis Data Model) framework offers a structured approach for the development of analysis datasets that are prepared for subsequent statistical analysis. The ADaM framework establishes standardized structures and variables for various types of analyses, including efficacy and safety assessments [62]. The proper formatting of analysis datasets is ensured, encompassing essential variables, derived variables, and statistical flags. The utilization of ADaM facilitates the streamlining of the analysis procedure, enabling statisticians to allocate their attention to the analysis properly, rather than expending time and effort on data preparation. In addition, ADaM enhances the ability to track and comprehend statistical analyses by providing a well-defined and documented structure and content for the analysis datasets.
One notable benefit of utilizing SDTM and ADaM is the enhanced efficacy observed in regulatory submissions. Regulatory bodies, such as the U.S. Food and Drug Administration (FDA), highly advocate for the adoption of standardized data models such as the Study Data Tabulation Model (SDTM) and the Analysis Data Model (ADaM). By adhering to these models, sponsors can furnish regulatory authorities with data that is systematically structured, uniform, and readily comprehensible. This accelerates the process of reviewing and allows regulators to more effectively assess the effectiveness and safety of novel treatments, resulting in prompt approvals and expedited access to new therapies for patients. In addition, the adoption of SDTM (Study Data Tabulation Model) and ADaM (Analysis Data Model) promotes cooperation and facilitates the exchange of data among members of the scientific community. The effectiveness of data exchange and result comparison among various researchers from various organizations can be enhanced through the utilization of standardized models for data organization. This practice facilitates transparency and expedites the advancement of scientific knowledge by enabling others to verify and expand upon preexisting research outcomes [64]. Moreover, the extensive implementation of SDTM and ADaM facilitates the creation of analytical tools and software that align with these models, thereby augmenting the efficiency and efficacy of statistical analysis in the context of clinical trials. The utilization of the Study Data Tabulation Model (SDTM) and the Analysis Dataset Model (ADaM) offers significant value in the context of statistical analysis within clinical trials. These models play a crucial role in maintaining the coherence, comparability, and interpretability of data, thereby facilitating efficient analyses, streamlined regulatory submissions, and improved collaboration among members of the scientific community [65,66]. The utilization of SDTM (Study Data Tabulation Model) and ADaM (Analysis Data Model) enhances the reliability and replicability of statistical analyses, thereby playing a pivotal role in the progression of medical understanding and the enhancement of patient welfare.
There are no human subjects in this article and informed consent is not applicable.
Sagarkumar Patel and Srinivasa Reddy conceived the presented idea and designed the analysis. Also, both carried out the Reviews and wrote the manuscript with support from Rachna Patel and Sandeep Bolla. All authors discussed the results and contributed to the final manuscript. All authors read and approved the final manuscript.
I would like to express my very great appreciation to the co-authors of this manuscript for their valuable and constructive suggestions during the planning and development of this research work.
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.