Macrophages are key players in inflammation and atherosclerosis. They express surface receptors of different subtypes for the endogenous autocoid adenosine. Macrophages within atherosclerotic lesions attain two clear-cut functional phenotypes M1 (pro-inflammatory) and M2 (immunosuppressive). This study examines the relative expression of adenosine receptors and proteins involved in cholesterol transport in THP-1 human macrophages upon differentiation into M1 and M2 subtypes.
THP-1 human monocytes were cultured in the presence of 100nM PMA. When a differentiated non-polarized phenotype (M0) was achieved, cells were incubated in the presence of 20ng/ml interferon-?+100ng/ml LPS to obtain M1 macrophages or 20ng/ml IL-4 to obtain M2 subset. Phenotypes were confirmed via QRT-PCR and by flow cytometry. Expression of cholesterol efflux proteins (ABCA1, ABCG1, SR-B1 and 27 hydroxylase) and scavenger receptors (CD36, SR-A1, LOX1 and CXCL16) was analyzed by QRT-PCR and confirmed by Western blot.
The M1 subset of macrophages display reduced expression of cholesterol efflux proteins: ABCA1, SR-B1 and 27 hydroxylase, as compared to M0 and M2. However, expressions of SR-B1 and 27-hydroxylase (27OH) are lower in both M1 and M2 when compared to not polarized M0 subset. Moreover, M2 polarized macrophages display an increased expression of the major scavenger receptors: CD36, SR-A1 and LOX1. This provides an explanation for significantly higher internalization of oxLDL and foam cell formation in M2 versus M1. Our results demonstrate that the M1 phenotype is associated with upregulation of the A2AR and A2BR while the M2 phenotype displays enhanced A1 and A3R expression.
Adenosine, Inflammation, Atherosclerosis, Cholesterol transport
ABC transporters: ATP-Binding Cassette Transporters, CD36: Cluster of Differentiation 36, CXCL16: Chemokine CXC Ligand 16, CXCL10: Chemokine CXC Ligand 10, CCL17: Chemokine CC Ligand 17, CCR7: Chemokine CC Receptor 7, LPS: Lipopolysaccharide, GAPDH: Glyceraldehyde-3-Phosphate Dehydrogenase, LDL: Low-Density Lipoprotein, LOX1: Lectin-like Oxidized Low-Density Lipoprotein Receptor, PMA: Phorbol 12-Myristate 13-Acetate, RCT: Reverse Cholesterol Transport, SR-A1: Scavenger Receptor Class A Type 1, SR-B1: Scavenger Receptor Class B Type 1, 27OH – 27-Hydroxylase.
Inflammation is the reaction of the immune system to infection or injury and has been implicated in the pathogeneses of multiple diseases such as arthritis, cancer, and stroke, as well as neurodegenerative and cardiovascular disorders [1-4]. Intrinsically, inflammation is a beneficial event that leads to restoration of tissue structure and physiological functions. The macrophage is an integral part of innate and adaptive immunity and is composed of two subtypes that are either pro-inflammatory or anti-inflammatory [5]. The classically activated or M1 macrophage phenotype produces inflammatory cytokines, like tumor necrosis factor (TNF)-a, that trigger acute inflammation. The alternatively activated M2 phenotype is anti-inflammatory and has been found to play an important role in tissue and wound repair [6,7]. Although the usual outcome of the acute inflammatory response is successful resolution and repair of tissue damage, dysfunction may predispose to autoimmunity, chronic inflammation, and accumulating tissue damage [8,9]. Over-production of M1 pro-inflammatory macrophages has also been linked to the development of atherosclerosis [10,11].
Stress and inflammation cause an interruption in oxygen supply (i.e., hypoxia) and accumulation of extracellular adenosine [12-14]. Adenosine signaling pathways are activated via one of its specific G-coupled protein receptors: A1R, A2AR, A2BR, and A3R. There is growing evidence that the function of the adenosine receptors is crucial in an anti-inflammatory response, reducing ischemia-reperfusion injury in multiple organs [15-18]. Previous studies demonstrate that A2AR and A3R activation inhibit the nuclear factor (NF)-?B signaling pathway, consequently triggering the transcription of the TNF-a gene [19,20]. Hence, A2AR and A3R agonists are proposed as candidates for anti-rheumatic agents due to their ability to reduce expression of TNF-a [20,21].
Anti-inflammatory properties of A2AR and A2BR agonists are associated with their anti-atherogenic capabilities, as indicated by previous studies performed by our group [22,23] and others [24]. The anti-atherogenic effect of activation of A2AR is attributed to its enhancement of reverse cholesterol transport by upregulating cholesterol 27-hydroxylase and the ATP binding cassette transporter A1 (ABCA1), a transporter that mediates cholesterol efflux to lipid-poor apolipoproteins [25].
This study defines a link between changes in expression of adenosine receptors and cholesterol handling in polarized THP-1 human macrophages. Our results demonstrate that the M1 phenotype is associated with upregulation of the A2AR and A2BR while the M2 phenotype displays enhanced A1 and A3R expression. Accordingly, the M1 phenotype amasses less oxLDL than the M1 phenotype, which corresponds to reduced foam cell formation.
THP-1 monocytes were obtained from the American Type Culture Collection (Rockville, MD). Cell culture media and supplementary reagents were obtained from Invitrogen (Carlsbad, CA).
Phorbol 12-myristate 13-acetate (PMA) and Oil red O were purchased from Sigma-Aldrich (St. Louis, MO). The selective A2AR antagonist 4-(2-[7-amino-2-(2-furyl) [1,2,4]triazolo[2,3-a] [1,3,5]triazin-5-ylamino]ethyl)phenol (ZM-241385) was purchased from from Abcam Inc. (Cambridge, MA). The selective A2AR antagonist 8-[(E)-2-(3,4-Dimethoxy-phenyl)-vinil]-1,3_diethyl-7-methyl-3,7-dihydro-purine-2,6-dione (KW-6002) was obtained from Axon Medchem (Groningen, Netherlands).
Trizol reagent was purchased from Invitrogen (Carlsbad, CA). All reagents for reverse transcription-polymerase chain reaction (RT-PCR) were purchased from Applied Biosystems (Foster City, CA). SYBR Green Master mix for the quantitative real-time polymerase chain reaction (QRT-PCR) was obtained from Roche Applied Science (Penzberg, Germany). Primers used in amplification reactions were generated by Sigma-Genosys (St. Louis, MO).
Cell culture media and supplementary reagents were obtained from Invitrogen (Carlsbad, CA). The THP-1 human monocytic leukemia cell line was chosen for the study because it shares many properties with normal human monocytes, including expression of scavenger receptors and cholesterol transport proteins, and is a well-accepted model for atherosclerosis used in our laboratory [26,27] and many others.
THP-1 monocytes (American Type Culture Collection, Manassas, VA) were grown at 37oC in a 5% CO2 atmosphere in RPMI 1640 supplemented with 10% fetal calf serum (FCS), 50 units/ml penicillin, and 50units/ml streptomycin. To facilitate differentiation into macrophages, THP-1 monocytes were treated with 100nM PMA for 24h at 37°C. When a differentiated non-polarized phenotype (M0) was achieved, the PMA-containing medium was removed, and replaced with complete RPMI 1640 supplemented with 10% FCS for another 24 hours.
In order to generate polarized phenotypes, cells were then cultured for 48 hours in the presence of 20ng/ml interferon-? and 100ng/ml LPS to obtain M1 macrophages [28,29] or 20ng/ml IL-4 to obtain M2 macrophages [30]. Phenotypes were confirmed via QRT-PCR analysis of gene expressions in commonly used cell surface markers IL-12, CCR7, and CXCL10 for M1 and CD163, mannose receptor, and CCL17 for M2 [31,32] (Table 1).
Table 1: Macrophage phenotype markers used for QRT-PCR. View Table 1
Phenotypical changes in macrophages were verified by flow cytometry using a BD Accuri C6 flow cytometer (BD Biosciences, Franklin Lakes, NJ). Both M1 and M2 phenotypes were stained with a PE conjugated mAb to CD163, a FITC conjugated mAb to CD68, an APC-conjugated mAb to CD11b, and a PE/Cy7 conjugated mAb to CD14, as well as isotype matched control mAbs, for 30 minutes at room temperature according to the manufacturer’s instructions (Abcam, Cambridge, MA) [31-33].
Immediately after the incubation period, total RNA was isolated with Trizol (Carlsbad, CA) reagent (1ml/106 cells). The quantity of total RNA from each condition was measured by absorption at 260 nm and 280nm wavelengths by ultraviolet spectrophotometry (Hitachi U2010 spectrophotometer).
ABCA1, ABCG1, 27-hydroxylase, SR-B1, PPAR gamma, LXR alpha, LOX1, SR-A1, CXCL16, CD36, A1R, A2aR, and A3R mRNA were quantified by real-time PCR. The cDNA was copied from 1 µg of total RNA using Moloney murine leukemia virus reverse transcriptase primed with oligo dT. Equal amounts of cDNA were taken from each RT reaction mixture for PCR amplification using specific primers (Table 2). QRT-PCR analysis was performed using the SYBR Green Reagent Kit according to the manufacturer's instructions on the Roche Light Cycler 480 (Roche Applied Science, Penzburg, Germany). Each reaction was done in triplicate. The amount of PCR products were estimated using Roche Applied Science software, provided by the manufacturer. Fluorescence emission spectra were monitored and analyzed. PCR products were measured by the threshold cycles (CT), at which specific fluorescence becomes detectable. The CT value for each gene was normalized to that of glyceraldehydes-3-phosphate dehydrogenase (GAPDH), and the relative expression level was calculated as the mean value of the untreated THP-1 as 1. Non-template controls were included for each primer pair to check for significant levels of any contaminants. A melting-curve analysis was performed to assess the specificity of the amplified PCR products.
Table 2: The list of specific primers used for QRT-PCR. View Table 2
Cellular extracts were prepared with Lysis Kit - RIPA Buff¬er (ProteinSimple, Santa Clara, CA). The immunoreactive proteins were detected using Wes Assay kit (ProteinSimple, Santa Clara, CA).
Rabbit anti-human ABCA1 (sc-20794) (Santa Cruz, CA) and rabbit anti-human ABCG1 (ab-36969) (Abcam Inc., Cambrige, MA) were used as primary antibodies for detection of ABCA1 and ABCG1, respectively. Rabbit anti-human LOX1 (ab60178), CD36 (ab64014), SR-A1 (ab36625), CXCL16 (ab101404) were purchased from Abcam Inc. (Cambrige, MA). As a loading control, GAPDH was detected using rabbit anti-human GAPDH antibody (ab9485).
Quantization of detected proteins and image preparation were performed with Compass Software (ProteinSimple, Santa Clara, CA).
For the foam cell formation assay, cells were washed in distilled water and stained with 0.2% Oil Red O (Sigma, St. Louis, MO) for 30 min. After the PBS wash, cell nuclei were stained with hematoxylin (Sigma, St. Louis, MO) for 5 min. After a final wash with PBS, coverslips were mounted on slides using Permount solution (Sigma, St. Louis, MO). Foam cells, recognized as red-stained cells, were visualized via light microscopy (Axiovert 25; Carl Zeiss, Gottingen, Germany) with 40x magnification and photographed using a DC 290 Zoom digital camera (Eastman Kodak, Rochester, NY). The number of foam cells formed in each condition was calculated in triplicate manually and presented as percentage of total cells.
Cells were incubated with 5 µg/ml 1,1'-dioctadecyl-3,3,3',3'-tetramethylin docarbocyaninet (DiI)-oxLDL (Intracel, Frederick, MD) for 4h. Slides were prepared using Vectashield mounting medium containing DAPI stain (Vector Laboratories, Inc., Burlingame, CA). After incubation, accumulation of DiI-oxLDL in cells was determined by fluorescent intensity with a Nikon A1 microscopy unit with 40X magnification and photographed with a DS-Ri1 digital camera. Fluorescent intensity was quantified from at least 3 random fields (1024x1024 pixels) per slide, from 3 slides per experimental condition and graphed.
Statistical analysis was performed using Graphpad Prism, version 5.01 (GraphPad Software, San Diego, CA). All data were analyzed by one way ANOVA with a Bonferroni post hoc test to evaluate the statistical significance of intergroup differences in all the tested variables. P values < 0.05 were considered statistically significant.
M1 macrophages express IL-12, CCR7, and CXCL10 at significantly higher levels than M2 macrophages (P< 0.001, n=3) (Figure 1A-C). M2 macrophages express CD163, mannose receptor, and CCL17 at significantly higher levels than M1 macrophages (P< 0.001, n=3) (Figure 1D-F).
 Figure 1: Surface marker expression on THP-1 monocytes, M0 and on M1 and M2 macrophage subsets Total RNA was isolated from monocytes and macrophages of each phenotype (M0=checkered; M1=horizontal stripes; M2=vertical stripes), reverse transcribed and amplified by PCR with GAPDH message as an internal standard. Gene expression levels were graphed as fold difference in mRNA expression versus monocytes set as 1.0. The data represent the mean + SEM of 3 independent experiments. M1 markers: (A) IL-12 expression (B) CXCL10 expression (C) CCR7 expression; M2 markers (D) CD163 expression (E) Mannose Receptor (F) CCL17 expression.
Figure 1: Surface marker expression on THP-1 monocytes, M0 and on M1 and M2 macrophage subsets Total RNA was isolated from monocytes and macrophages of each phenotype (M0=checkered; M1=horizontal stripes; M2=vertical stripes), reverse transcribed and amplified by PCR with GAPDH message as an internal standard. Gene expression levels were graphed as fold difference in mRNA expression versus monocytes set as 1.0. The data represent the mean + SEM of 3 independent experiments. M1 markers: (A) IL-12 expression (B) CXCL10 expression (C) CCR7 expression; M2 markers (D) CD163 expression (E) Mannose Receptor (F) CCL17 expression.
(**) P< 0.01; (***) P< 0.001 vs. monocytes; (###) P< 0.001 vs. M2 macrophages; (&&&) P< 0.001 vs. M1 macrophages.
View Figure 1
Phenotypes were confirmed via flow cytometric analysis of standardized cell surface markers. Mean fluorescent intensities were analyzed to confirm M1:M2 polarization.
M1 polarized macrophages display reduced expression of the major cholesterol efflux proteins at levels that significantly differ from the expression profile in M2 polarized macrophages (Figure 2). Thus, level of ABCA1, SR-B1 and 27 hydroxylase (27OH) were 0.54 + 0.052, 0.22 + 0.04 and 0.15 + 0.05 lower than non-polarized M0 macrophages (P< 0.001, n=3). The level of ABCG1 was not significantly different (0.90 + 0.06) upon activation of M1 phenotype compared to M0 and M2 (1.17 + 0.016) (Figure 2B). In contrast, M2 macrophages exhibit upregulation in the expression of ABCA1 (1.49 + 0.003) (P< 0.001, n=3) (Figure 2A). Even though mRNA levels of SR-B1 and 27OH were significantly higher in M2 polarized macrophages versus M1, their levels were reduced as compared to M0 macrophages (Figure 2C, D): 0.54 + 0.013 and 0.38 + 0.015, respectively (P< 0.001, n=3) . Gene expression analysis was confirmed by Western blot (Figure 2E, F).
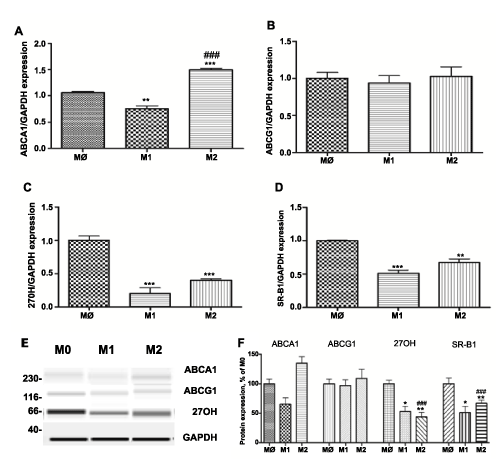 Figure 2: Expression of cholesterol efflux proteins: ABCA1, ABCG1, SR-B1 and 27-hydroxylase (27-OH) in different subsets of THP-1 macrophages.
Figure 2: Expression of cholesterol efflux proteins: ABCA1, ABCG1, SR-B1 and 27-hydroxylase (27-OH) in different subsets of THP-1 macrophages.
Gene expression was evaluated in each subset of macrophages (M0 – naïve differentiated with PMA, M1 – IFN/LPS stimulated, and M2 – IL4 stimulated) using equal amounts of cDNA. mRNA levels were graphed as fold difference in mRNA expression versus M0 macrophages set as 1.0. The data represent the mean + SEM of 3 independent experiments. (A) ABCA1 expression (B) ABCG1 expression (C) 27OH expression and (D) SR-B1 expression (E) Western Blot for ABCA1, ABCG1 27OH, and GAPDH protein expression (F) Relative protein expression levels of ABCA1, ABCG1 27OH, and SR-B1 in M0, M1, and M2 macrophages.
(*) P< 0.01; (**) P< 0.01; (***) P< 0.001 vs. M0 macrophages; (###) P< 0.001 vs. M1 macrophages.
View Figure 2
M1 differentiated macrophages express reduced levels of scavenger receptors: CD36, SR-A1 and LOX1 compared to M2 macrophages (Figure 3). Levels of CD36 and LOX1 were significantly lower: 0.84 + 0.009 and 0.54 + 0.004, respectively, compared to M0 non-polarized macrophages. In contrast, expression of CXCL16 was 10 fold higher compared to M0 and M2 macrophages (P< 0.001, n=3) (Figure 3D). M2 polarized macrophages express 2 fold higher levels of CD36 and SR-A1 (P< 0.011, n=3) and 4 fold higher LOX1 (P< 0.011, n=3) (Figure 3 A-C). The level of CXCL16 was not affected by polarization to M2 phenotype (Figure 3D). Gene expression analysis was confirmed by Western blot (Figure 3E,F).
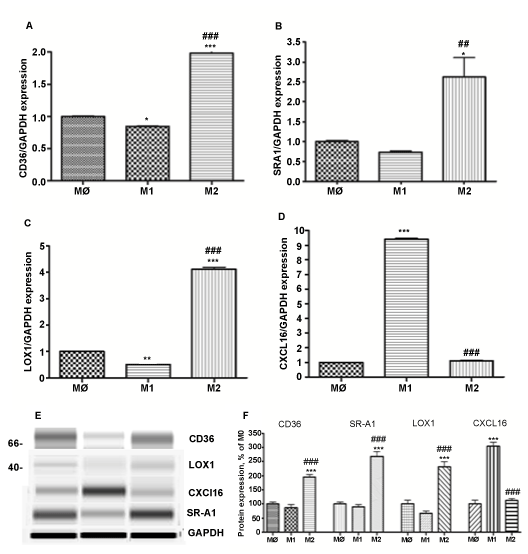 Figure 3: Expression of scavenger receptors: CD36, SR-A1, LOX1 and CXCL16 in different subsets of THP-1 macrophages.
Figure 3: Expression of scavenger receptors: CD36, SR-A1, LOX1 and CXCL16 in different subsets of THP-1 macrophages.
Gene expression was evaluated in each subset of macrophages (M0 – naïve differentiated with PMA, M1 – IFN/LPS stimulated, and M2 – IL4 stimulated) using equal amounts of cDNA. mRNA levels were graphed as fold difference in mRNA expression versus M0 macrophages set as 1.0. The data represent the mean + SEM of 3 independent experiments. (A) CD36 expression (B) SRA1 expression (C) LOX1 expression (D) CXCL16 expression (E) Western Blot for CD36, SRA1, LOX1, CXCL16, and GAPDH protein expression (F) Relative protein expression levels of CD36, SRA1, LOX1, and CXCL16 in M0, M1, and M2 macrophages.
(*) P < 0.01; (**) P < 0.01; (***) P < 0.001 vs. M0 macrophages; (###) P < 0.001 vs. M1 macrophages.
View Figure 3
In our study, M1 polarized macrophages uptake 1.5 times less DiI-oxLDL than M2 differentiated subset (P< 0.001, n=3), as presented on microphotograph (x40x) (Figure 4A). Fluorescent intensity was quantified from at least 3 random fields (1024x1024 pixels) per slide, from 3 slides per experimental condition and graphed (Figure 4B). Lower lipid accumulation correlated with decrease in the foam cell formation in M1 versus M2 macrophages (Figure 4A). Consequently, M2 macrophages formed 25.5 + 5.4% (P< 0.001, n=5) more foam cells as compared to M1 subset (Figure 4C). Number of foam cells formed in each condition was calculated in triplicate manually and presented as percentage of total cells.
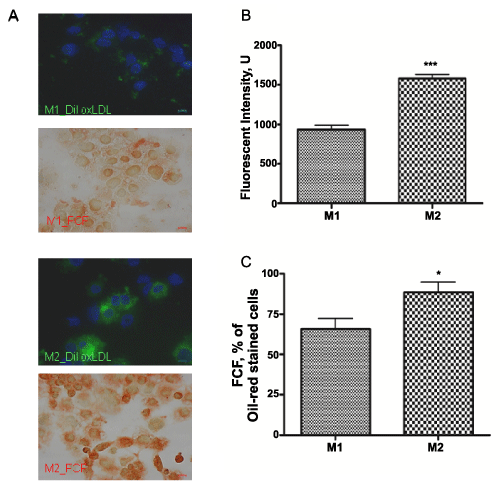 Figure 4: Internalization of DiI-oxLDL and foam cell formation in M1 and M2 THP-1 macrophages.
Figure 4: Internalization of DiI-oxLDL and foam cell formation in M1 and M2 THP-1 macrophages.
Cells were photographed with a DS-Ri1 digital cameras (A) Integrated DiI-oxLDL are presented in green, DAPI nuclear staining – in blue and foam cells were recognized by Oil-red-O staining. oxLDL internalization was measured as fluorescent intensity of integrated DiI-oxLDL and graphed (B) Foam cell formation (FCF) was calculated as a percentage of oil-red-O stained cells (C) All results are expressed as means + SEM of three independent experiments. (*) P< 0.01; (***) P< 0.001 for M1 vs. M2 macrophages.
View Figure 4
Upon polarization, M1 macrophages express 2.5 fold and 3 fold increased A2AR and A2BR, respectively (P< 0.011, n=3) and a reduced amount of A3R (P< 0.001, n=3) (Figure 5B-D) compared to M0 and M2 subsets. In contrast, M2 macrophages display reduced level of A2A (0.53 + 0.022; P< 0.001, n=3) and significantly not different level of A2B (Figure 5B,C). M2 polarized macrophages express 1.5 fold increased A1R (P< 0.001, n=3) and 2.2 fold higher A3R (P< 0.001, n=3) (Figure 5A,D) compared to M0 and M1 macrophages.
 Figure 5: Expression of adenosine receptors: A1, A2A, A2B and A3 in THP-1 macrophage subsets. Gene expression was evaluated in different subsets of macrophages (M0=naïve differentiated with PMA, M1=IFN/LPS stimulated, and M2=IL4 stimulated) using equal amounts of cDNA. mRNA levels were graphed as fold difference in mRNA expression versus M0 macrophages set as 1.0. The data represent the mean + SEM of 3 independent experiments. (A) A1R expression (B) A2AR expression (C) A2BR expression (D) A3R expression. (**) P< 0.01; (***) P< 0.001 vs. M0 macrophages; (###) P< 0.001 vs. M1 macrophages.
View Figure 5
Figure 5: Expression of adenosine receptors: A1, A2A, A2B and A3 in THP-1 macrophage subsets. Gene expression was evaluated in different subsets of macrophages (M0=naïve differentiated with PMA, M1=IFN/LPS stimulated, and M2=IL4 stimulated) using equal amounts of cDNA. mRNA levels were graphed as fold difference in mRNA expression versus M0 macrophages set as 1.0. The data represent the mean + SEM of 3 independent experiments. (A) A1R expression (B) A2AR expression (C) A2BR expression (D) A3R expression. (**) P< 0.01; (***) P< 0.001 vs. M0 macrophages; (###) P< 0.001 vs. M1 macrophages.
View Figure 5
Macrophages promote atherogenesis by internalizing modified lipoproteins that have penetrated the vessel wall to form cholesterol-loaded foam cells. The phenotypic switch in macrophages present in atherosclerotic lesions influences disease development and progression [34,35]. Multiple studies have attempted to solve the M1/M2 macrophage polarization paradigm. It is generally accepted that the pro-inflammatory population is pro-atherogenic while the anti-inflammatory subset is atheroprotective [36,37]. However, M1 and M2 macrophages both contribute during diverse stages of human plaque development and localize in distinct morphological points within atherosclerotic lesions. Further, the clearcut difference between M1 and M2 macrophages in vitro is probably not the natural circumstance. Degree of differentiation toward one state or the other probably varies along a spectrum. Thus, evaluation of molecular and biochemical changes upon their differentiation is valuable to our understanding of atheroma progression.
In this study we demonstrate that the M1 phenotype expresses low levels of the proteins involved in cholesterol efflux – ABCA1 and SR-B1 as compared to M2 macrophages. These results are in accord with previous reports by Waldo et al, (2008) and Bouhlel et al, (2007) [34,38]. We did not observed changes in the expression of ABCG1 in M1 and M2 subsets, compared to [34], and down regulation of 27-hydroxylase, a protein that provides a pathway for elimination of intracellular cholesterol [39], in both subsets compared to the M0 phenotype. These findings authenticate pro-atherogenic properties in the M1 subset of macrophages. However, expression of SR-B1 and 27-hydroxylase are lower in both M1 and M2 when compared to undifferentiated M0 macrophages (Figure 2). Moreover, M2 polarized macrophages display an increased expression of the major scavenger receptors responsible for modified lipid uptake: CD36, SR-A1 and LOX1. This supports the current model for increased lipid uptake in M2 macrophages as reported in Canton et al, (2013) [40]. In combination with the decreased level of cholesterol efflux proteins, this provides an explanation for observed difference in the internalization of oxLDL: significantly higher in M2 versus M1 (Figure 4). Lipid accumulation, in turn, leads to elevated foam cell production [40], observed in the M2 population in our study. Interestingly, for the M1 set of macrophages, expression of only one scavenger receptor was increased - chemokine CXC ligand 16 (CXCL16) – a novel scavenger receptor, involved in oxLDL uptake [41].
Despite documented differences in behavior of human and murine macrophages, a preponderance of our data regarding macrophage polarization originates in murine studies, while information on human macrophages remains sparse [42]. Further, the importance of adenosine receptors in atherosclerosis progression has been explored in murine models and not evaluated properly in humans. Thus, in the present study, conducted in a human cell line, we observed that M1 polarization is associated with upregulation of the A2AR and A2BR while M2 polarization enhances A1 and A3R expression.
While studies have shown possible beneficial effects of A2AR activation on the survival rate in septic murine models, recent evidence has shown that pharmacologic blockade of the A2AR using selective antagonists such as ZM-241385 decreases mouse mortality under septic conditions [17]. As these data occur in septic/inflammatory models where the distribution of polarized macrophages is shifted toward the M1 phenotype, future studies are indicated to further elucidate adenosine receptor activity in both M1 and M2 macrophages. Determining the effects of A2AR inhibition on macrophage polarization will also add to our understanding of the complicated physiology of adenosine action.
The clinical relevance of the present work is highlighted by the ongoing Cardiovascular Inflammation Reduction Trial (CIRT), an NIH-sponsored randomized, double blind, placebo-controlled clinical trial designed to determine whether low dose methotrexate will reduce recurrent cardiovascular events in 7,000 stable patients who are post-myocardial infarction or have documented coronary artery disease with type 2 diabetes or metabolic syndrome [43]. Atheroprotective effects of methotrexate are mediated, at least in part, through elevation of adenosine levels and thus the impact of methotrexate on the cardiovascular health of these patients may be dependent on macrophage phenotype in their arterial plaques [44]. Other potential approaches to controlling macrophage cholesterol accumulation may involve dietary supplements such as resveratrol which has low toxicity and may act on cholesterol transport proteins through adenosine or other signaling pathways [45,46] Given the contrasting properties of pro-inflammatory M1 and anti-inflammatory M2 macrophages in homeostasis and disease, considering the balance between these populations has been deemed crucial in developing therapeutic remedies.
This work was supported by R21 AT007032-01A1 from The National Center for Complementary and Alternative Medicine and by the Elizabeth Daniell Research Fund.