Insulin resistance is a major player in the pathogenesis of type 2 diabetes. C3 converts to C3a and acylation stimulating protein (ASP) by complement activation. ASP activates adipose tissue macrophages and accelerates continuous inflammation. Tissue-bound C3a is known to induce insulin resistance. We examined the relationship between insulin resistance and complement activation by the in vivo alteration of ASP. The levels of ASP and parameters of insulin resistance and inflammation were measured in plasma from 25 patients with type 2 diabetes. A selective peroxisome proliferator-activated receptor gamma (PPAR) agonist was administered to these 25 patients for three months, and then ASP and the parameters were measured again. The alterations of ASP and the parameters were examined in the pathophysiological state. Plasma levels of ASP and complement activation products were significantly higher in diabetes. Parameters of insulin resistance were also significantly increased. The ASP level significantly improved after PPAR agonist administration. The improvement in ASP level correlated with improvements in parameters of insulin resistance and inflammation. The levels of other relevant adipocytokines by insulin resistance correlated with the degree of inflammation. ASP and complement activation were associated with insulin resistance and chronic inflammation in diabetes.
Acylation stimulating protein, Adipocytokine, Complement activation, Diabetic nephropathy, Insulin resistance, Chronic inflammation
Insulin resistance is responsible for the metabolic disorders in patients with type 2 diabetes, and plays a role in the expression of vascular complications [1]. Insulin resistance causes hyperinsulinemia, resulting in the establishment of vascular injury by accelerating vascular smooth muscle proliferation [2] and vascular endothelial dysfunction [3]. However, the underlying etiology of insulin resistance in diabetes is complex and seems to be multifaceted.
Recently, the contribution of the complement system to insulin resistance was elucidated [4,5]. Complement proteins are known as adipocytokines secreted from adipocytes as well as hepatocytes, and play a role in the pathophysiological state of obesity and dyslipidemia as well as in the host-defense system [6]. The amplification circuit of the alternative complement pathway is continuously activated in the postprandial hyperlipidemic state, and persistent complement activation would induce atherosclerotic plaque formation [7]. In patients with obesity with dyslipidemia, complement proteins oversecreted from active adipocytes are gradually activated by chylomicron. C3 is converted slowly but persistently to C3a and C3b [8]. C3b binds to Factor B and to another circulating C3 again to form C5 convertase. This complex activates the membrane attack pathway to form membrane attack complex (MAC) and atherosclerotic plaque subsequently. C3a immediately loses its anaphylatoxin activity to form C3a desArg or acylation stimulating protein (ASP) in the fluid phase [9]. Mamane et al. [10] reported that the C3a receptor was a key mediator of insulin resistance and functions by modulating adipose tissue macrophage infiltration and activation in the rodent model. Type 2 diabetes patients and obese patients with hypercomplementemia and insulin resistance have a high incidence of renal injury and cardiovascular events [11]. Liver-derived complement components would also be activated in chylomicron-rich circumstances and would amplify insulin resistance. In this study, the authors investigate the relationship between insulin resistances and complement activation products such as ASP.
Twenty-five over weight patients (mean ± SD: 68 ± 11 years of age; body mass index, BMI>24.0kg/m2) who were previously diagnosed with and were being treated for type 2 diabetes and hyperlipidemia (total cholesterol >250mg/dL before treatment) were enrolled in this clinical study (diabetes group). The duration of diabetes was 10.8 ± 6.7 years (mean ± SD). Those with serum creatinine >2.0mg/dl were excluded to avoid the influence of renal filtration failure. Patients undergoing lipid-lowering therapy using statins or undergoing anti-hypertensive therapy using angiotensin II receptor 1 blocker (ARB) continued their therapy as usual throughout this study. Calcium channel blockers or diuretics were used in 16 of the 25 patients. Sulfonylureas were used in 18 patients, and biguanides were used in 23 patients. Other medicines that alter glycemic control were not used. Twenty-two over-weight volunteers without diabetes and hypertension (mean ± SD: 63 ± 17 years of age) were selected as the control group (Table 1,2). In the diabetes group, according to the urinary albumin/creatinine ratio (ACR), nine patients had macroalbuminuria (over 300mg/g•Cr), 11 patients had microalbuminuria (more than 40mg/g•Cr, but less than 300mg/g•Cr), and five patients had normoalbuminuria (less than 40mg/g•Cr). The diabetes group also underwent retinal assessment; six patients had proliferative retinopathy, 11 patients had preproliferative retinopathy, and 8 patients had background retinopathy. Other clinical background data are shown in Table 1.The clinical background of the patients with type 2 diabetes such as glycemic control, obesity, and insulin resistance was characteristic of that of ordinary patients with type 2 diabetes.
Table 1: Clinical characteristics of 25 patients with type 2 diabetes in the baseline. View Table 1
Table 2: Plasma levels of ASP and other parameters in 22 healthy controls and 25 patients with type 2 diabetes. View Table 2
Plasma levels of C3, its activation intermediates such as iC3b, ASP, and membrane attack complex (MAC), adipocytokines such as adiponectin (ADN) and tumor necrosis factor (TNF), and other lipid mediators such as low density lipoprotein-cholesterol (LDL-C), high density lipoprotein-cholesterol (HDL-C), and triglyceride (TG) were measured in the 25 patients with type 2 diabetes. The same parameters were measured in the 22 healthy control subjects. All patients agreed to participate in this study and to have peroxisome proliferator-activated receptor gamma (PPAR treatment. Pioglitazone, 15 mg per day, was administered to the diabetes patients for 3 months to improve their insulin resistance. Post-treatment measurements were carried out at the end of the study, and prepared for the control. The levels of complement activation were examined and compared between the different states of insulin resistance using PPAR agonist. Ethylenediaminetetraacetic acid (EDTA)-containing plasma samples were collected at the time the patient visited our hospital and were stored at –80°C until use. Urine was collected by the patients early in the morning and brought to the hospital on the same day, and the urine samples were also stored at –80°C until use. The data were compared between pre- and post-study. The influence of PPAR improvement on adipocytokine excretion, complement activation, and renal vascular injury was examined. This study was approved by the IRB committee of our hospital. All subjects agreed to participate in this study at the time of sample collection.
Complement activation products, comprising iC3b, ASP, and soluble MAC (sMAC), were measured by ELISA at our laboratory. Assay kits were purchased from BD Biosciences (San Jose, CA, USA) for ASP, and Quidel Corporation (San Diego, CA, USA) for iC3b and sMAC. Plasma levels of high-molecular-weight adiponectin (ADN) and tumor necrosis factor alpha (TNF) were also measured by ELISA at our laboratory. Assay kits for ADN (Fujirebio Inc., Tokyo, Japan) and TNF Matriks Biotechnology Ltd Sti, Ankara, Turkey) were purchased. Other parameters were measured at the central laboratory department of our hospital: plasma glucose by GOD-enzyme electrode assay; immuno-reactive insulin by electrochemiluminescence immunoassay (ECLIA); HbA1c by the latex agglutination method; C3 by laser nefelometry; creatinine by enzymatic assay; and urinary albumin by immunoprecipitation. The value for HbA1c was revised by the National Glycohemoglobin Standardization Program (NGSP) after measurement by the previous Japanese standard substance and assay. Homeostasis model assessment of insulin resistance (HOMA-IR) was calculated by fasting plasma glucose and immuno-reactive insulin level, estimated glomerular filtration rate (eGFR) was calculated by serum creatinine, body mass index (BMI) was calculated by body length and body weight, and urinary albumin creatinine ratio (ACR) was calculated by urinary albumin and creatinine.
All values are presented as the mean ± standard deviation for continuous variables. Mann-Whitney U test was used for the comparisons, and Spearman rank coefficient was used for correlations. A p value of < 0.05 was considered statistically significant. Calculations were performed with commercial software, JMP ver.6.0.3 (SAS Institute Japan, Tokyo, Japan).
The plasma level of ASP was significantly higher in the baseline samples from the 25 patients with type 2 diabetes (224 ± 36µg/mL) than in the 22 healthy controls (169 ± 22µg/mL).Complement component and complement activation products were also significantly higher in the patients with type 2 diabetes (C3: 121 ± 37mg/dL vs. 92 ± 22mg/dL, iC3b: 306 ± 21μg/mL vs. 65 ± 27μg/mL, sMAC: 3.43 ± 0.70ng/mL vs. 2.13 ± 0.72ng/mL) (Table 2).
The relationships between ASP and related parameters were examined. ASP was significantly correlated with HOMA-IR and IRI (HOMA-IR: R2=0.3153, p=0.0257; IRI: R2=0.3357, p=0.0171) (Figure 1). BMI was correlated with HOMA-IR and serum triglyceride (TG) was correlated with IRI(HOMA-IR: R2=0.3750, p=0.0073; TG: R2=0.3370, p=0.0167 (Figure 1). These data indicate that obesity and dyslipidemia are associated with insulin resistance and hyperinsulinemia, which would be associated with the excess levels of plasma ASP. Plasma sMAC was correlated with disease duration (R2=0.1026, p=0.0233). The levels of TNF and ADN, which were known to be secreted as well as C3 from adipocytes in the accelerated insulin-resistant state, were correlated with glomerular dysfunction (TNF vs. eGFR: R2=0.6003, p=0.0001; ADN vs. eGFR: R2=0.3779, p=0.0068).
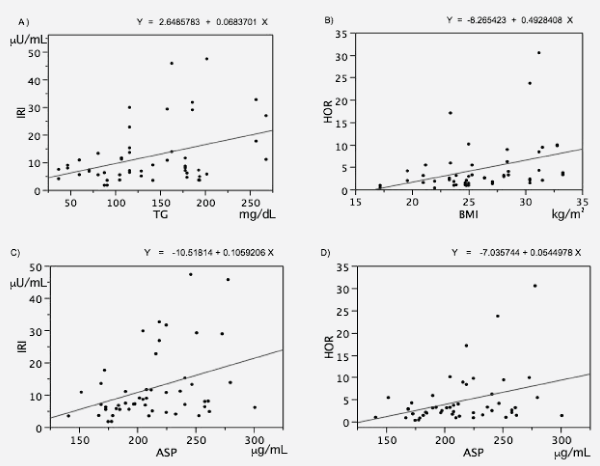 Figure 1: Correlations between acylation stimulating protein (ASP) and other parameters of insulin resistance. (A) Serum triglyceride (TG) correlates with immuno-reactive insulin (IRI). (B) Body mass index (BMI) correlates with homeostasis model assessment of insulin resistance (HOR). (C), (D) ASP has a significant correlation with HOR and IRI. These data suggest that obesity and dyslipidemia are associated with insulin resistance and hyperinsulinemia, which are associated with the overproduction of ASP.
View Figure 1
Figure 1: Correlations between acylation stimulating protein (ASP) and other parameters of insulin resistance. (A) Serum triglyceride (TG) correlates with immuno-reactive insulin (IRI). (B) Body mass index (BMI) correlates with homeostasis model assessment of insulin resistance (HOR). (C), (D) ASP has a significant correlation with HOR and IRI. These data suggest that obesity and dyslipidemia are associated with insulin resistance and hyperinsulinemia, which are associated with the overproduction of ASP.
View Figure 1
After treatment with 15mg/day of Pioglitazone for three months, plasma ASP significantly improved from 224 ± 36μg/mL to 203 ± 35μg/mL, and HOMA-IR also improved from 6.5 ± 7.4 to 2.8 ± 2.2, accompanying the improvement of insulin resistance. The levels of sMAC, C3, and iC3b significantly improved from 3.43 ± 0.70ng/mL to 2.91 ± 0.54ng/mL, from 121 ± 37mg/dL to 101 ± 32mg/dL, and from 306 ± 21μg/mL to 254 ± 25μg/mL, respectively. The direct correlations were recognized between the markers of insulin resistance (HOMA-IR) and that of inflammation (ASP, sMAC). These data suggest that a relationship between improvement of PPAR by Pioglitazone may related with the improvement of complement activation and/or diabetic vascular injury. There were no correlations between other parameters (Table 3). The close relationship between ASP and insulin resistance was also revealed in human diabetic pathophysiology, and not only in model animals.
Table 3: Alterations of plasma ASP and other parameters after improvement of insulin resistance with PPAR treatment. View Table 3
The authors examined the plasma levels of ASP, other complement activation products, and parameters of insulin resistance in 25 patients with type 2 diabetes. We prospectively examined the alteration of this cohort along with the improvement of insulin resistance by PPAR agonist treatment. We found a close relationship between complement activation and insulin resistance in our small size cohort.
Complement components are overproduced from active adipocytes and activated in the presence of chylomicron in patients with obesity with dyslipidemia [12]. C3 is then converted to C3a and C3b in the amplification circuit of the alternative complement pathway [13]. C3b binds to Factor B and another C3 to form C5 convertase which activates the membrane attack pathway. C3a immediately loses its anaphylatoxin activity to form ASP.C3a and ASP have distinct receptors in the target organs, and accelerate insulin resistance and inflammation independently [14,15]. Tissue-bound C3a was reported by Mamane et al. [10] as an accelerator of insulin resistance in a rodent model. Cianflone et al. [16,17] reported that C3a and ASP have distinct receptors, and that the C3a receptor would accelerate insulin resistance and inflammation and ASP receptor would accelerate lipid metabolism. However, Munkonda et al. [18] reported that administration of recombinant ASP accelerated insulin resistance. Therefore, the effect of ASP on insulin resistance remains somewhat controversial. It is difficult to examine the distinct effects of C3a and ASP independently because C3a in the circulation is immediately inactivated and converted to ASP. In this study, it was difficult to clarify which of C3a and ASP would increase in the circulation, and play a role in the development of insulin resistance. C3a and ASP would bind to the target tissues through distinct receptors, and would play roles in insulin resistance and inflammation, which are different from their roles in the fluid phase. Immediate inactivation of C3a and the existence of distinct receptors for C3a or ASP lead to difficulty in clarifying the mechanisms of signal transmission after the binding to each receptor. An additional limitation to the present study includes the relatively small size of subjects. Further studies are necessary to clarify this issue.
The known accelerating factors of insulin resistance are listed as hypertension, hyperglycemia, hyperlipidemia, obesity etc., [19,20]. Adipocytokines are associated with each pathophysiological condition, and would accelerate the formation of vascular complications. The effect of C3a or ASP on insulin resistance is of value in considering the expression of vascular complications in type 2 diabetes patients [21]. We showed in this study that plasma C3, iC3b, ASP, and sMAC levels were significantly higher in diabetes patients in comparison with healthy controls. These data would suggest over-secretion of adipocytokines from adipocytes and the persistent activation of complement pathways in patients with type 2 diabetes. The correlations between HOMA-IR and ASP or BMI or TG indicate that complement activation would attend diabetic pathophysiology through insulin resistance in patients with obesity with dyslipidemia [19,22]. Although insulin resistance is associated with complement activation in this study, it remains unclear which state would induce the other condition. However, previous reports suggested that complement activation products induce insulin resistance in the rodent model [9,10]. Activation of complement needs the background of obesity and dyslipidemia [13], and it is common in patients with type 2 diabetes or metabolic syndrome. In the circumstance of high plasma C3a, the acceleration of inflammation would contribute to organ injury in patients with type 2 diabetes. Even liver-derived complement components would be activated in chylomicron-rich circumstances and would also amplify insulin resistance and glomerular dysfunction. Plasma TNF and adiponectin were correlated with eGFR in this study, and this suggests that these adipocytokines are associated with the expression of organ injury and vascular complications through complement activation and inflammation. Proinflammatory cytokines activate adipose tissue macrophages and induce chronic inflammation, resulting in organ injury [23]. Insulin resistance and adipocytokines play roles in the expression of diabetic macroangiopathy and microangiopathy [24]. Insulin resistance may also induce endothelial dysfunction, resulting in the expression of vascular complications [25,26]. Endothelial dysfunction in renal arterioles is known as one of the causes of microalbuminuria and glomerular dysfunction [27,28]. These markers of glomerular dysfunction were correlated with adipocytokines in this study, which suggests that complement activation causes insulin resistance and inflammation, leading to the expression of vascular complications such as organ injury and atherosclerosis.
Aadipocytokines and proinflammatory cytokines are increased in plasma from overweight patients with type 2 diabetes with dyslipidemia. Excess C3a and ASP would bind to the target tissues and accelerate continuous inflammation and insulin resistance in organ. Therefore, persistent complement activation would accelerate vascular complications in association with insulin resistance.
This work was supported by the constitutional subsidy from Nihon University School of Medicine, Nihon University Itabashi Hospital, and Nihon University Medical Alumni Association. All authors have no potential conflict of interests to declare associated with this manuscript.