Good's syndrome (GS) that consists of hypogammaglobulinemia and thymoma is a rare adult-onset disease. Some paraneoplastic syndromes like pure red cell aplasia (PRCA) and myasthenia gravis (MG) may accompany to this immunodeficiency disease. We herein reported two fatal cases of GS.
Case 1 describes a 65-year-old man with GS who applied with the symptoms of immunodeficiency six months after a treatment including thymectomy and radiotherapy. Medical history, clinical findings and laboratory results of the patient were consistent with GS. The patient received IVIG treatment regularly during the 5 years without any infection. Then PRCA, digital enfarcts and sepsis developed consecutively and he died due to sepsis when he was 72 years old. Case 2 describes a 39-year-old man with thymoma and MG. He was administered radiotherapy, chemotherapy and thymectomy. He was diagnosed as GS when he was admitted to hospital due to drug-resistant pneumonia one year after the thymectomy. IVIG treatment wasn't administered, because clinical and hemodynamic status of the patient worsened due to sepsis in a short time and he died of respiratory and cardiac arrest.
In conclusion, there are so many clinical presentations of immune deficiency in adults. GS is one the late onset immunodeficiency diseases of the adults. Regarding to this, the patients with thymoma should be evaluated with respect to immune deficiency both before and after thymectomy.
Thymoma, Hypogammaglobulinemia, Pure red cell aplasia, Immunodeficiency, Good's syndrome
Thymoma is one of the most common mediastinal neoplasms with 20% ratio and a wide variety of paraneoplastic disorders including myasthenia gravis, pure red cell aplasia (PRCA), and so forth can accompany to it. PRCA and hypogammaglobulinemia occurs in a significant proportion of patients in the presence of thymoma (5-15% and < 5% respectively) [1]. Good's syndrome (GS) that is a rare disorder of the immune system involving B and T cells and characterized by thymoma usually occurs in the 4th or 5th decade of life [2,3]. We herein reported two fatal cases of GS; one of them presented with acquired PRCA and digital necrosis, and the other one presented with congestive heart failure and pneumonia.
A 65-year-old male patient was diagnosed thymoma incidentally after a traffic accident and undergone thymectomy and radiation therapy in 2007. Six months after the surgery, he was referred to our clinic due to a prolonged febrile illness with recurrent vesiculo-ulcerative lesions at his oral mucosa. Medical history, clinical findings and laboratory results of the patient were consistent with the diagnosis of GS. Serum protein electrophoresis showed decreased albumin, elevated α globulin and depressed δ globulin peaks (Albumin: %46.9 (54.6-66), α1-globulin: %14.4 (1.4-2.8), α2-globulin: %17.9 (9.1-13.8), β-globulin: %15.2 (8.7-14.4), γ-globulin: %4 (10.6-19.2)) and the levels of major serum immunoglobulin isotypes were found lower than their normal ranges ( IgG: 5.91g/L (7-16), IgA: < 0.25g/L (0.7-4), IgM: 0.17g/L (0.4-2.3)). Lymphocyte subset analysis revealed a decreased ratio of CD4+ T cells to CD8+ T cells due to a decreased percentage of CD4+ T cells and an increased percentage of CD8+ T cells, and diminished percentage of CD19+ B cells (CD45+: %92.3 (88-100), CD14+: %5.2 (2-8), CD3+: %81.5 (62.8-85), CD4+: %21.3 (34-63.8), CD8+: %50.1(19-48), CD19+: %0.1 (7-23), CD19+/CD27+: %0, CD4+/CD8+: %0.42 (0.8-3.3), CD3-/CD16+/CD56+: %6.4(6-29)). IVIG treatment was administered at a dose of 600 mg/kg IV and maintained once every three weeks and subsequently a crucial improvement was seen both in clinical findings and laboratory results.
After a regular 5 years follow up period with IVIG treatment, the patient applied to our clinic with the complaints of weakness, loss of appetite and weight loss in January 2012. A complete blood count showed that white blood cell count (WBC): 2.1x103/mm3 (4.3-10.3), hemoglobin (Hb): 1.3g/dl (13.6-17.2), hematocrit (Hct): 9.7 % (39.5-50.3) and platelet count (Plt): 356x103/mm3 (156-373). The patient was hospitalized due to severe anemia. Peripheral blood smear and bone marrow aspirate specimen smear revealed hypochromic microcytic anemia and a reduction in erythroid cells respectively and subsequently bone marrow biopsy was also examined by a hematopathologist: Bone marrow was normocellular (50% cellularity) with intact granulocytic and megakaryocytic elements, and erythroid colonies were virtually absent. Immunperoxidase staining for glycophorin highlited randomly distrubuted, non-colonized erythroid precursors. Giant erythroblasts and parvoviral intranuclear inclusions were not determined. Scattered reactive T-lymphocytes were present in bone marrow but not any reactive B-lymphocyte. Bone marrow biopsy showed that histological and immunohistochemical findings were compatible with PRCA (Figure 1a-1d). An intensive tranfusion treatment was performed and Hb value increased as 9.9g/dl and Hct value increased as 9.7%. But eight days later a febrile respiratory distress and cyanosis of the fingers except the left hand occurred (Figure 2a,2b). Computed tomography was performed and a broad-spectrum antibiotherapy, intravenous immunoglobullin M (pentaglobin) and vasodilatator treatment were administered with the diagnosis of sepsis.
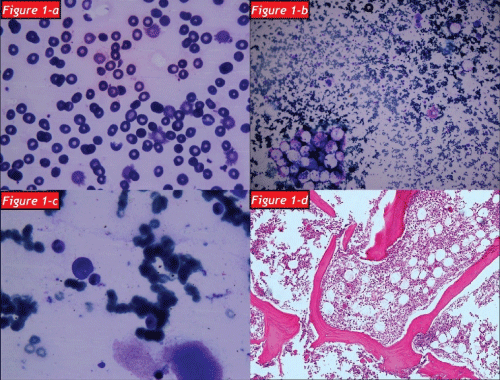 Figure 1: The blood smear shown that thrombocytopenia, hypochromic microcytic aemia and schistocytes
Figure 1: The blood smear shown that thrombocytopenia, hypochromic microcytic aemia and schistocytes
Figure 1b: The bone marrow aspiration shown that myeloid and erythroid dysplasia
Figure 1c: Micromegakaryocytoid forms
Figure 1d: Bone marrow section with paucity of erythroid elements (H&E x100).
View Figure 1
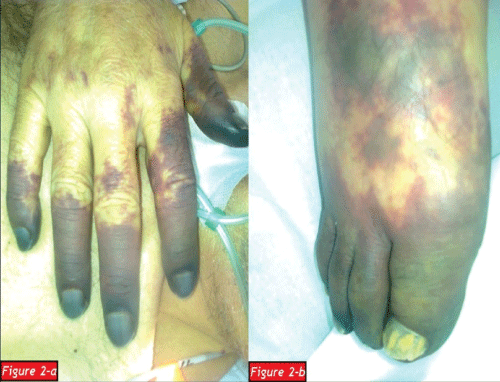 Figure 2a: Cyanotic fingers of the right hand
Figure 2a: Cyanotic fingers of the right hand
Figure 2b: Cyanotic fingers of the right foot.
View Figure 2
At the twentieth day of treatment the infectious status regressed, but circulatory disorder converted to digital infarcts. The patient was consulted by cardiovascular surgery and orthopedy clinics and cyanotic distal phalanx of the fingers were amputated. Two days later, clinical status of the patient began to deteriorate all of a sudden. Despite to all lifesaving treatments orientation, cooperation and hemodynamics got worse and the patient died of respiratory and cardiac arrest due to disseminated intravascular coagulation and pulmonary embolism when he was 72 years old.
A 39 year-old male patient was diagnosed as thymoma in 1997 and myasthenia gravis (MG) in 2000 respectively. He was administered radiotherapy in 1997, chemotherapy in 2004 and undergone thymectomy in 2011 for the treatment of thymoma. He received deflazacort and pyridostigmin for the treatment of MG between 2000 and 2012. One year after the thymectomy he applied to emergency service with the complaints of dispnea, loss of appetite and bilateral lower extremity edema. He was hospitalized due to congestive heart failure and pneumonia. Postero-anterior chest radiography and high-resolution computed tomography (HRCT) of the thorax were reported as opportunistic infection (Figure 3a- 3d) Sputum cultures yielded acinetobacter baumanii and cytomegalovirus. He was consultated by immunology and allergy service due to febrile drug-resistant pneumonia. Medical history, clinical findings and laboratory results of the patient were determined to be compatible with the diagnosis of GS.
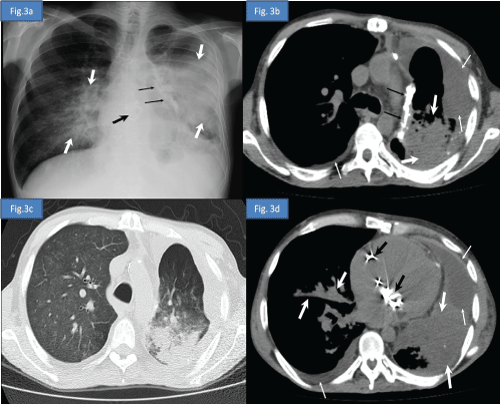 Figure 3a: Postero-anterior chest radiography demonstrates decreased volume of the left hemithorax, bilateral heterogeneous consolidation areas more prominent in left lung secondary to pneumonic infiltration (white thick arrows), left mediastinal pleural calcified plaque (black thin arrows), prosthetic cardiac valves (black thick arrow) and obliteration of the left costodiaphragmatic sinus due to pleural effusion.
Figure 3a: Postero-anterior chest radiography demonstrates decreased volume of the left hemithorax, bilateral heterogeneous consolidation areas more prominent in left lung secondary to pneumonic infiltration (white thick arrows), left mediastinal pleural calcified plaque (black thin arrows), prosthetic cardiac valves (black thick arrow) and obliteration of the left costodiaphragmatic sinus due to pleural effusion.
Figure 3b: Axial computed tomography image at the upper thoracic level in soft tissue window and in lung window.
Figure 3c: Axial computed tomography image at the lower thoracic level and in soft tissue window
Figure 3d: Axial computed tomography image show decreased volume of the left lung, bilateral pleural effusion (thin white arrows), prosthetic cardiac valves (black thick arrow), left mediastinal pleural calcified plaque (black thin arrows), bilateral heterogeneous consolidation areas more prominent in left lung and extensive ground-glass opacities in right lung secondary to pneumonic infiltration (white thick arrows).
View Figure 3
A depressed gamma globulin peak was observed in serum protein electrophoresis (gamma-globulin: %7.6). The levels of major serum immunoglobulin isotypes were decreased ( IgG: 4.7g/L, IgA: 0.576g/L, IgM: < 0.175g/L). A diminished percentage of CD19+ and CD20+ B cells were found in lymphocyte subset analysis (CD45+: 96.9%, CD14+: 9.8%, CD3+: 84.1 %, CD19+: 0.1%, CD20+: 0.4%, CD22+: 6.9%, CD24+: 0.3%, surface CD79A+: 0.3%, cytoplasmic CD79A+: 1.6%, CD4-+CD8+: 21.1%, CD4++CD8-: 15.1%, CD4++CD8+: 40.4%, CD3-/CD16+/CD56+: %6.3). IVIG treatment wasn't administered because clinical and hemodynamic status of the patient worsened due to sepsis in a short time. The patient reacted unresponsive to antimicrobial therapies and lifesaving treatments, and died of respiratory and cardiac arrest at the third day of hospitalization.
GS that first reported by Good in 1954 is one of the rare causes of primary immune deficiencies (PID) of adults. Combined B and T cell immunodeficiency (hypogammaglobulinemia, diminished B cells, reduced CD4+/CD8+ T cell ratio) and thymoma are the main characteristics of this syndrome [4].
PRCA that primarily inhibits erythropoiesis may occur either congenital or acquired [5] secondary PRCA may accompany thymoma, malignant lymphoma, chronic leukemia, systemic lupus erythematosus and Sjögren's syndrome [6]. Association of GS and PRCA was reported in a few case reports [1,4,6-10]. The common features of these cases were clinical findings related to low levels of immunoglobulins and PRCA occurred after thymectomy like this patient [1,8].
Treatment of thymoma primarily involves the surgical removal of tumor but radiotherapy and chemotherapy are also necessary in patients with advanced stage tumors [7]. Treatments for thymoma patients with PRCA and immunodeficiency are various. The complete PRCA remission rate after thymectomy is about 30% [11]. Besides, immunosuppressive treatments and intravenous immunoglobulin replacement treatments are needed to perform a complete remission and reduce infections [11]. But improvement in the management of GS is diverse and usually the prognosis is quite poor [2]. Individuals with GS are prone to be ill especially caused by opportunistic infectious agents. Infectious, autoimmune and hematologic complications are the main causes of death in GS.
Our first case diagnosed as GS when he was 65-year-old. He had chronic drug resistant oral mucosa lichen planus and candidiasis history which was compatible with an immune deficiency disease. His clinical findings manifested after the removal of thymoma. Defective immunoglobulin production, lack of mature B cells and impaired cellular immunity were found at immunological analysis. Although his clinical status was improved owing to regular IVIG treatment, PRCA was deteriorated gradually. When PRCA was added to GS five years after the thymectomy any tumor recurrence or metastasis of the thymus was not determined. Our case was different from the other case reports of GS with his hematologic status. He was diagnosed as PRCA with the determination of a severe anemia which wasn't compatible with life even for more younger ages (Hb: 1.3 g/dl, Hct: 9.7% ) [1,6]. According to case reports in the literatüre the patients with GS and PRCA had a good survival related to their haemoglobin and immunoglobulin values [1,4,6-8]. But, unfortunately the patient in question did not have a similar survival due to sepsis. Also, circulatuar disorder which caused treatment-resistant digital necrosis worsened the clinical status. This situation also adds a privilege to our case report. Our second case also has an importance related to his age and clinical status. He was 39 year-old when diagnosed as GS. This is an early age to have GS when compared to other GS cases in the literature [1,4,6-8]. This case's clinical and hemodynamic status worsened very fastly, so we could not find enough time to administer IVIG therapy for the treatment of hypogammaglobulinemia.
In conclusion, there are so many clinical presentations in adults with PID. These patients may apply to various clinics. This disease may remain undiagnosed for a long time because of the lack of awareness. Especially the patients with recurrent, atypical and drug-resistant infections of the skin, respiratory and gastrointestinal tract, and the patients who have a family history of similar diseases should be considered to be a possible PID. Some autoimmune, allergic or malign diseases may play a key role in PID pathogenesis and this situation should be investigated as soon as possible. Also, the patients with thymoma should be evaluated with respect to immune deficiency both pre and post thymectomy.
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Informed written consent was obtained from the patient for the publication of this case report.