Purpose: To describe a case of atypical flattening of fovea centralis and corneal irregularity in whom a 7q11.23 deletion consistent with Williams-Beuren Syndrome.
Methods: This is an observational case report. The medical history of the patient was assessed. Ophthalmologic examination including slit-lamp biomicroscopy, dilated indirect ophthalmoscopy, fundus autofluorescence and optical coherence tomography (OCT) imaging, full-field electroretinography, multifocal electroretinography and corneal topography was performed and literature was reviewed.
Results: Previous genetic analysis (fluorescence in situ hybridization method) had confirmed the diagnosis of William-Beuren syndrome in this patient by detection of the deletion on chromosome 7q11.23. Visual acuity of the patient was 0.7 for both eye with Snellen chart. OCT imaging showed the flattening of the foveal contour and corneal topography revealed the finding of irregular corneal surface. All other ophthalmologic tests were normal.
Conclusion: The authors report the first case of a patient with ophthalmic findings characteristic for flattening of fovea centralis and corneal irregularity mimicking keratoconus in the setting of genetically confirmed Williams–Beuren syndrome.
Fovea, OCT, Cornea, Williams-Beuren syndrome
Williams-Beuren Syndrome (WBS) is a rare, mostly sporadic neurodevelopmental genetic disorder resulting from well-defined hemizygous microdeletion of 1.5 to 1.8 Mb on chromosome 7 (7q11.23) which contains approximately 28 genes, whose manifestations include unique collection of somatic, brain, and cognitive features which are observed in 95% of individuals [1]. It occurs in 1 per 7,500-20,000 births. The syndrome was first described in 1961 by Williams and collegues from New Zealand and in 1962 by Beuren and collegues from Germany independently [2]. The characteristic facial features include protuberance around the eyes, short nose, wide mouth, prominent lips, full cheeks and a small jaw. They can also have a short stature, sloping shoulders, long neck and limited movement in their joints [3]. Elastin gene mutation causes proliferation of the smooth muscle cells and fibroblasts and arterial flexibility diminishes with irregular organization of short elastic fibers [4].
According to current literature ophthalmologic findings shown in patients with WBS include strabismus (most commonly esotropia), stellate pattern of the anterior iris stroma (mostly in individuals with blue or hazel iris color), tortuosity of retinal vessels and lacrimal duct obstruction. In the literature cases with keratoconus, retinitis pigmentosa, rod-cone dystrophy have also been reported [5]. This is the first reported WBS case with atypical foveal flattening, decreased ILM-RPE thickness accompanying irregular corneal surface mimicking keratoconus.
An 11-years-old female patient was referred to the Ophthalmology Department of Istanbul Medipol University for visual examination. In the ophthalmologic examination the refractive error was +1.00 (-1.00 × 10) for the right eye and +1.25 (-0.75 × 155) for the left eye. Her visual acuity was 0.7 for both eyes with Snellen visual acuity test. In biomicroscopic examination iris had a stellate pattern which was a sign of WBS (Figure 1). Increased vascular tortuosity was observed in fundus examination (Figure 2). OCT imaging showed flattened foveal curvature (Figure 3) and thinning in ILM-RPE thickness. Subfoveal choroidal thickness in our case was 345 μm and 339 μm respectively. Corneal topography revealed the finding of irregular corneal surface mimicking keratoconus (Figure 4 and Figure 5).
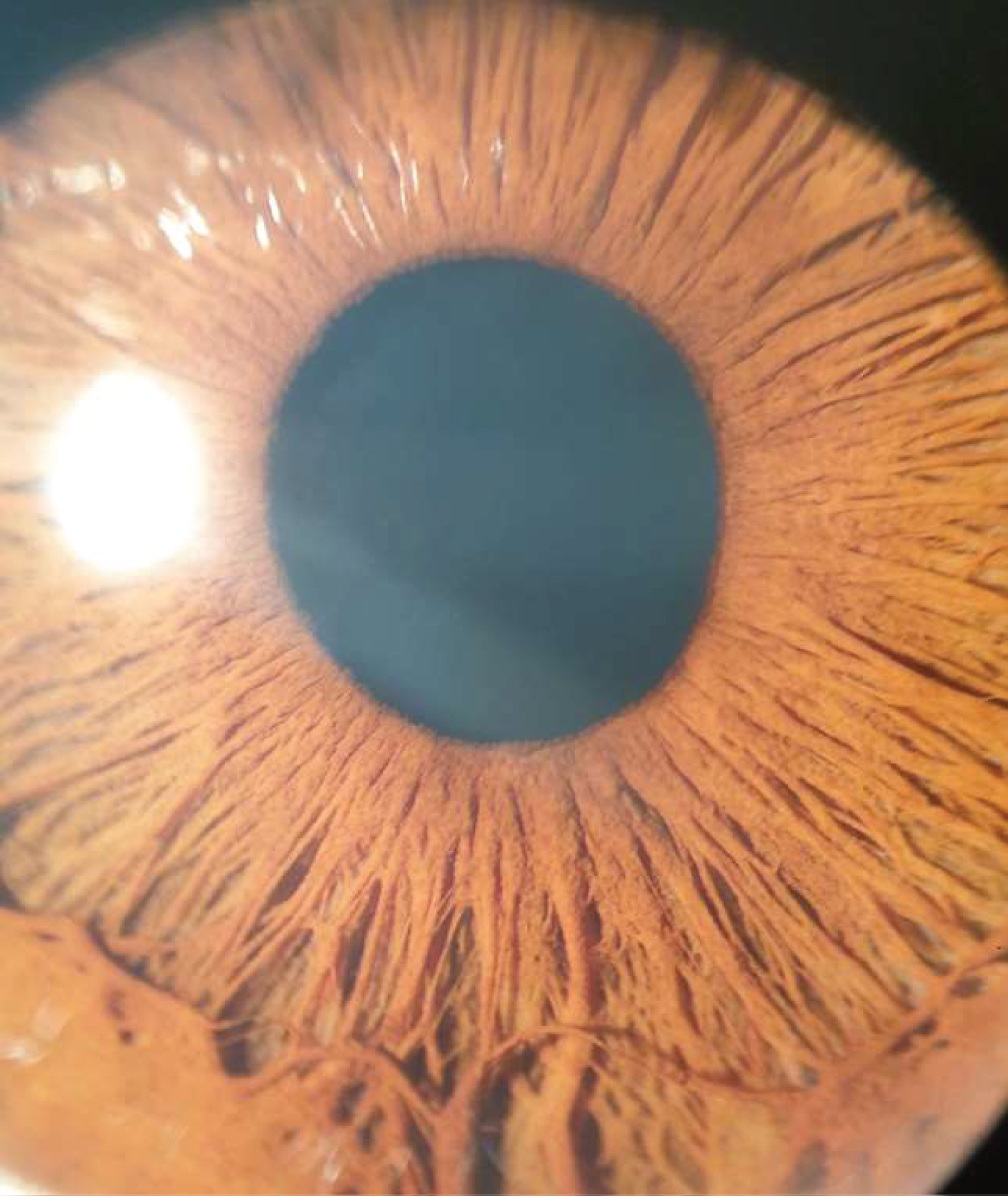 Figure 1: Stellate iris pattern of the patient.
View Figure 1
Figure 1: Stellate iris pattern of the patient.
View Figure 1
 Figure 2: Retinal vessel tortuosity.
View Figure 2
Figure 2: Retinal vessel tortuosity.
View Figure 2
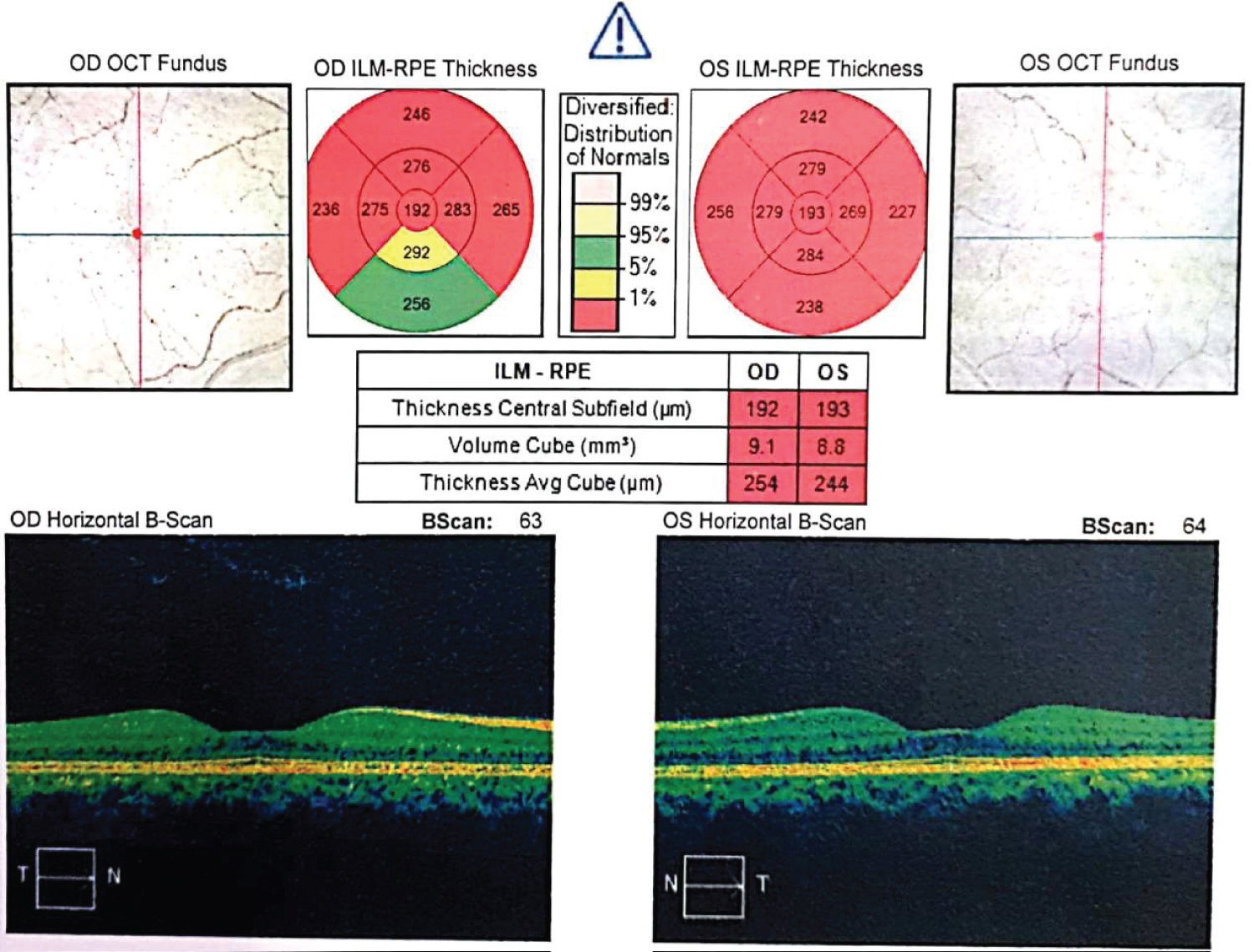 Figure 3: OCT imaging showed flattened foveal curvature and thinning in ILM-RPE thickness.
View Figure 3
Figure 3: OCT imaging showed flattened foveal curvature and thinning in ILM-RPE thickness.
View Figure 3
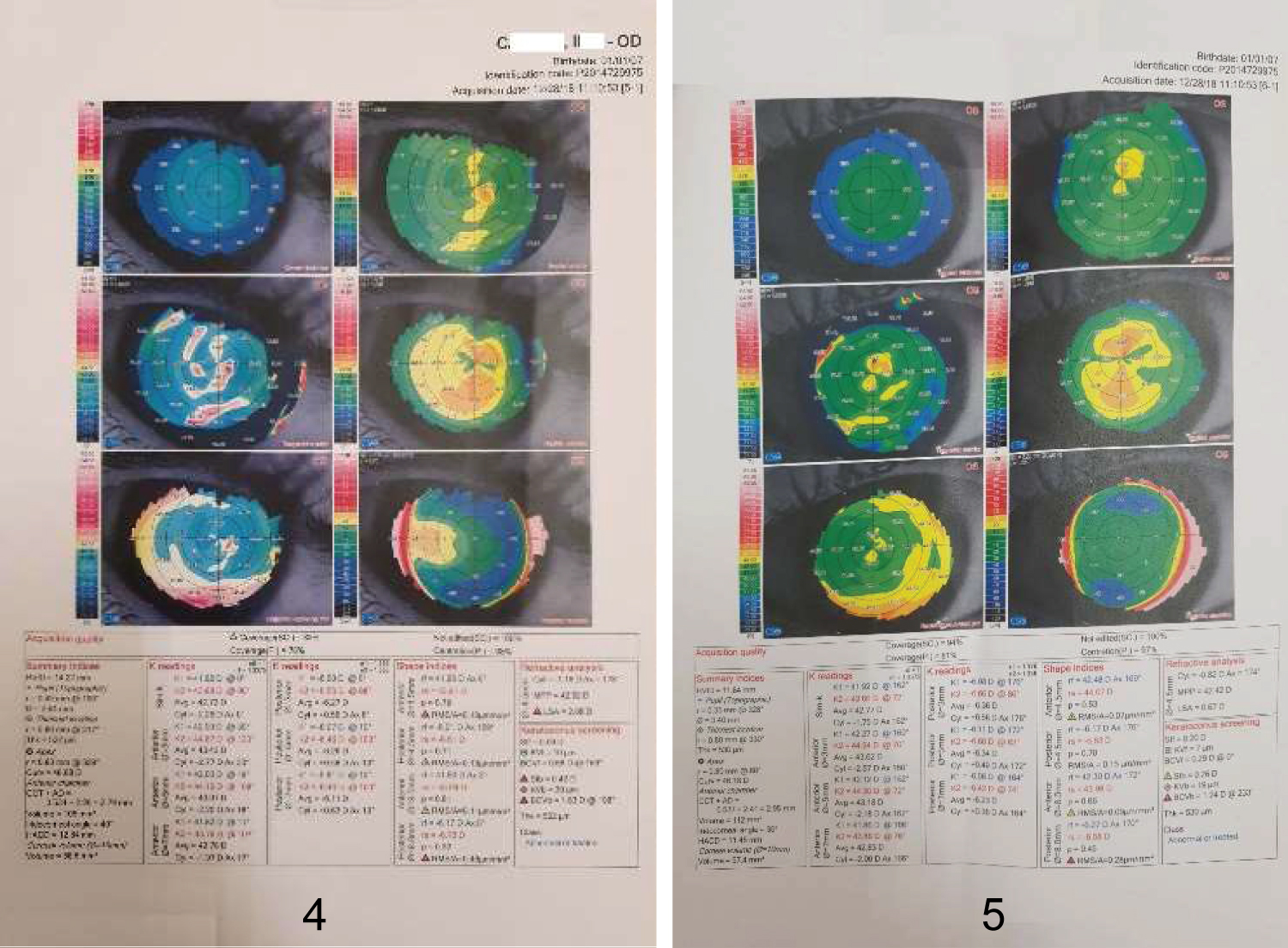 Figure 4 and Figure 5: Corneal topography revealed the finding of irregular cornea.
View Figure 4 & 5
Figure 4 and Figure 5: Corneal topography revealed the finding of irregular cornea.
View Figure 4 & 5
WBS is a multisystem disorder caused by the deletion of 26-28 contiguous genes, including elastin (ELN) gene on chromosome 7q11.23. It occurs in 1 per 7,500-20,000 births. Most of the cases show sporadic patterns [6]. In the eye, this gene was strongly expressed in the inner and outer nuclear layer of the retina [7]. This finding may be the reason of decreased ILM-RPE thickness seen on OCT imaging in this patient.
WBS patients typically have dysmorphic facial features including broad forehead, almond-shaped eyes, stellate pattern in the iris, high and prominent cheekbones, depressed nasal root, full lips, a broad mouth, dental malocclusion, small chin and prominent earlobes [8]. Our case also has stellate pattern of iris which stroma looks coarse with radial or cartwheel striations due to hypoplasia of iris stroma [3]. Holmstrom, et al. reported 51% of WBS patients had stellate pattern in the iris while 12% of the control subjects had this pattern. It was easier to detect in lightly pigmented iris [9].
Weber, et al. evaluated 30 children with WBS and they found that 77% of children had refractive error with the rate of 67% hyperopia, 20% astigmatism and 7% myopia [10]. Only one case of amblyopia was noted. 23% of children had epicanthus, and three children had stellate patterns of the iris. Eleven patients (36.6%) had strabismus, nine of them had esotropia, and two of them had exotropia. Binocular vision was abnormal in 43% of the patients. Diffuse arteriovenous tortuosity was seen in 27% of patients. Our case also had increased retinal tortuosity.
A case series of sixteen patients by Viana, et al. showed slightly higher rates of stellate irides (81.2%) and retinal vessel tortuosity (37.5%), but a lower rate of strabismus (18.7%). Patients with cataract (1/16), megalocornea (1/16), keratoconus (1/16) and optic disc hypoplasia (2/16) were also reported [11].
Kuehlewein, et al. have shown a case of rod-cone dystrophy associated with WBS (5). Their WBS subjects had overall preserved best-corrected distance visual acuity (VA) (8-9/10 ± 1.5 SD), which is consistent with previous reports and also apparently normal pattern of organization of the different retinal layers in all WBS participants [12]. Thinning in the outer retinal layer using optical coherence tomography and abnormalities in multifocal electroretinography have previously been reported in genetically confirmed thirteen WBS subjects with normal visual acuity and normal appearance of the macula and peripheral retina [10]. Castelo-Branco, et al. presented evidence of a neural defect in the retina of WBS patients. Decreased retinal thickness, abnormal optic disc concavity, and impaired visual responses have been reported with high-resolution imaging techniques in WBS patients compared to controls [12]. Subfoveal choroidal thickness in our case was 345 μm and 339 μm respectively which is thicker then normal. According to literature there is no information about this antity. Retinitis pigmentosa and WBS like features have been reported to be associated with myopathy and cerebellar ataxia only in two cases who are siblings. However a confirmed diagnosis of WBS by FISH test, was not made in these two cases [13]. In our case full field and multifocal electroretinogram tests were normal but OCT imaging showed flattened fovea, decreased ILM-RPE thickness with normal retinal segmentation. Flattened fovea might be associated with rod-cone dystrophy due to tight insertion of posterior hyaloid, but in our case because of electrophysiological tests were normal diagnosis of rod-cone dystrophy was excluded.
Mediero, et al. have shown the 4th case in the literature with WBS and keratoconus however their case was the first who undergone crosslinking treatment afterwards [14]. In this case is there is significant irregular astigmatism present (OD > OS) with some focal areas of steepening but the pattern is not diagnostic for keratoconus. Moreover, the posterior curvature does not show corresponding displacement nor pachymetric thinning in the areas of anterior steepening (which may indicate that the surface topography is being distorted by tear film or other surface pathology rather than from keratoconus). Lower SimKs and mild topographic cylinder also make the diagnosis of probable keratoconus questionable.
We report here the first case of a patient with ophthalmic findings characteristic decreased ILM-RPE thickness, abnormal flattening of fovea centralis resembling tight insertion of posterior hyaloid and mimicking rod-cone dystrophy pattern accompanying corneal surface irregularity in the setting of genetically confirmed WBS. These patients should be followed up closely by paediatric ophthalmologists for preventing visual disabilities as well as early detection of other complications in WBS.