Gyrate atrophy of the fundus is a rare autosomal recessive disease characterized by deficiency of ornithine-δ-aminotransferase. Ornithine-δ-aminotransferase deficiency causes hyperornithinemia which results in progressive chorioretinal atrophy. Plasma levels of ornithine are elevated. Based on these observations, we describe the diagnosis of a patient with gyrate atrophy of the choroid and retina. This is the first report of Gyrate atrophy in Cuba, diagnosed by ornithine levels in blood and the ophthalmologist assessment.
Gyrate atrophy, Hyperornithinemia, Electroretinogram, Retinal degeneration, Choroid, Pyridoxine
Gyrate atrophy (GA) of the choroid and retina (GACR) is an extremely rare inherited retinal dystrophy. The disease is an autosomic recessive disorder, caused by mutations in the ornithine aminotransferase gene (OAT), which is localized on chromosome 10q26 [1-4].
The gene is expressed in the neuroretina and in the retinal pigment epithelium (RPE) [5-7] and encodes for the mitochondrial and pyridoxal phosphate-dependent enzyme ornithine aminotransferase, which is necessary for the conversion of ornithine into glutamate and proline [8]. Consequently, OAT gene mutations result in hyperornithinemia, typically with a 10-20-fold elevation of plasma ornithine levels [9-12].
Hyperornithinemia and ornithinuria were not recognized as the biochemical counterparts of this disorder until the report of Simell and Takkin, in 1973, 85 years after the initial ophthalmologic description [13]. Several studies suggest that an arginine-restricted diet [14-25] or a low-protein diet [22-24] may slow the progression of chorioretinal lesions and visual loss in gyrate atrophy. Most patients seem not to be responsive to pyridoxine; however, a few studies report of pyridoxine-responsive patients presenting with a reduction of ornithine level after pyridoxine substitution [22-24].
The highest incidence has been described in Finland, where affects approximately 1 in 50,000 individuals. About 200 cases with biochemically confirmed GA have been reported in the literature (http://omim.org/entry/258870) [13].
The first symptoms of GACR patients are night blindness and constriction of the visual field, caused by sharply demarked round areas of chorioretinal atrophy in the periphery, which increase and coalesce over years and spread toward the macula resulting in visual loss [26,27] Additionally, myopia and early posterior subcapsular cataract are common in gyrate atrophy [28]. A further pathomorphological characteristic is cystoid macula edema, which can be detected in optical coherence tomography (OCT) scans [29,30].
The aim of this work is to describe the diagnosis of a first Cuban patient with gyrate atrophy of the choroid and retina through ophthalmological tests and biochemical results of the first case of gyrate atrophy with hyperornithinemia in Cuba.
Mixed 6-year-old female patient was referred to consultation because her teacher noted near-sightedness on school. The case was evaluated by the ophthalmology and genetics service. The patient was examined in 2014 and 2015 in the Department of neuro-ophthalmology by the "Ramon Pando Ferrer" Cuban Institute of Ophthalmology. Clinical genetics history was made, and plasma ornithine quantification was requested. Her family history was unremarkable. Clinical examinations and blood collection for serum ornithine level were conducted after obtaining informed consent. The research adhered to the tenets of the Declaration of Helsinki. The patient underwent a complete ophthalmological examination including best-corrected visual acuity (decimal fraction), slit-lamp biomicroscopy, and funduscopy. Kinetic visual field testing was performed with Goldmann, perimetry. OCT and FAF imaging were carried out using the Spectralis Heidelberg Retina Angiograph (Heidelberg Engineering, Heidelberg, Germany). For FAF, argon laser light (488 nm) was used to excite autofluorescence in the RPE, with a wide band-pass filter and a cutoff at 500 nm inserted in front of the detector. A 30_field of view was used to obtain mean images as provided during the imaging process.
The full-field electroretinogram (ERG) was recorded according to the International Society for Clinical Electrophysiology of Vision (ISCEV) standards [30,31] ERG recordings were made with a Nicolet Spirit and Ganzfeld bowl (Nicolet, Madison, USA) using DTL fiber electrodes (Unimed Electrode Supplies Ltd, England). Following 30 min of dark adaptation, four stimuli of increasing intensity were used to obtain rod responses and the standard combined rod-cone response (maximum light intensity: 1.5 cd s/m2).
After 10 min of light adaptation to a white background light of 25 cd/m2, single-flash cone and 30-Hz flicker responses were recorded using the maximum stimulus intensity.
A Prominence HPLC system (Shimadzu, Japan) is used for biochemical diagnosis of GACR. Gradient reverse-phase HPLC is used for quantification of amino acids in plasma with pre-column derivatization with phenyl isothiocyanate (PITC) and ultraviolet detection (254 nm). The LC Chromatography Data System Solutions program (Shimadzu, Japan) was used for data acquisition and processing. All determinations were performed at 30 ℃, using a Shim-pack ODS-GHRS 4 mm × 1 cm pre-column and Shim-pack CLC-ODS 25 cm, 5 µm column (Shimadzu, Japan), a flow of 1 mL/min and injection volume of 50 µL. Sample quantification relies on ornithine calibration curves prepared in plasma with concentration range of 100-800 µM.
Samples were processed as described by Camayd, et al. [32] using fresh random urine samples (2 mL or more). Briefly: Volumes corresponding to 0.25 mg of creatinine were alkalinized to pH = 14 and treated with 500 µL of hydroxylamine hydrochloride. The resulting mixture was acidified to pH = 1, saturated with approximately 500 mg of sodium chloride and extracted twice with ethyl acetate. Finally, the resulting organic phases were pooled, evaporated under Nitrogen at 50 ℃ and derivatized with 100 µL of N-Methyl-N-(trimethylsilyl)trifluoroacetamide. One microliter of each derivatized sample was injected. A GC-MS Shimadzu QP2010 equipped with an AOC-20i autoinjector (Tokyo, Japan) was used to carry out the analysis. Compounds were separated as trimethylsilyl derivatives using a DB-5 MS capillary column (30 m × 0.25 mm I.D., cross-linked 5% phenyl-methyl silicone, 0.25 µm film thickness). Chromatographic conditions were as follows: Helium was used as carrier gas (flow-rate of 1 mL/min), and split ratio was 1:10. The GC oven temperature was set from 80 ℃ (5 min hold) to 300 ℃ at a rate of 7 ℃/min (5 min hold). The interface and injector temperatures were 280 ℃ and 250 ℃, respectively, electron impact ionization applied was 70 eV. The mass spectrometer was programmed from a mass/charge ratio (m/z) 10-650 at the rate of 0.6 Hz.
To define pyridoxine responsiveness, the patient was treated with pyridoxine (500 mg/day) immediately after diagnosis for six months, and a follow-up sample was requested for amino acid analysis. Finally, a low-protein diet-supplemented with pyridoxine (500 mg/day)-was started and plasma amino acid levels were quantified after a six-month period of treatment and one year after diagnosis [22-24].
Mixed 6-year-old was referred to consultation because her teacher noted near-sightedness on school. The general ophthalmologist exam noted changes on fundus examination, suggestive of retinal dystrophy. Her refractive error measured -6.00 - 1.00 × 180° for the right eye and -6.50 - 1.00 × 35° for the left eye. Her best corrected visual acuity was 20/20 in the right eye and 20/25 in the left eye. Color vision was within normal limits in both eyes. Pneumotonometry revealed normal intraocular pressure (IOP) in each eye. Pupil reflex were normal. Anterior segment examinations were quiet bilaterally. Fundus examination revealed bilateral sharply demarcated areas of choroid and retinal atrophy in gyrate shape and involving the midperiphery, the posterior pole was spared of these lesions with the macula not affected. (Figure 1) Disclosed cystoid macular edema was evident in both eyes on optical coherence tomography (Figure 2). Electroretinography (performed to International Society for Clinical Electrophysiology of Vision standards) showed virtually abolished responses in scotopic conditions. 32 Dynamic Octopus perimetry demonstrated visual field constriction in each eye. (Figure 3) Electromyography was normal.
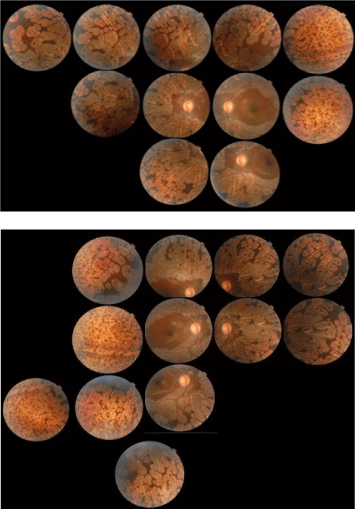 Figure 1: Fundus Photography showing bilateral sharply demarcated areas of choroid and retinal atrophy in gyrate shape involving the midperiphery, posterior poles were spared of these lesions.
View Figure 1
Figure 1: Fundus Photography showing bilateral sharply demarcated areas of choroid and retinal atrophy in gyrate shape involving the midperiphery, posterior poles were spared of these lesions.
View Figure 1
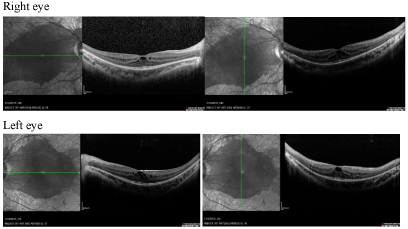 Figure 2: Optical coherence tomography showing thickened macula due to cystoid macular edema evident in both eyes. View Figure 2
Figure 2: Optical coherence tomography showing thickened macula due to cystoid macular edema evident in both eyes. View Figure 2
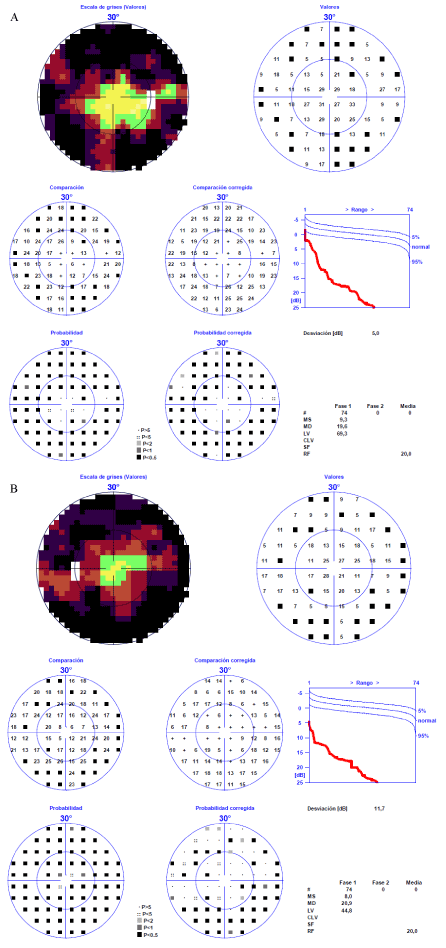 Figure 3: HAAG STREIT Octopus 101 (32 Dynamic) visual field testing A) Left eye and B) Right eye showing constricted fields. View Figure 3
Figure 3: HAAG STREIT Octopus 101 (32 Dynamic) visual field testing A) Left eye and B) Right eye showing constricted fields. View Figure 3
The patient was referred to the genetics service and clinical genetics history is made. Family history that proves to be the only case in the family, no positive perinatal history and there is a history of language disorders and learning. Amino acid analysis revealed a high plasma ornithine level demonstrating deficiency of the activity of OAT (Table 1). Plasma ammonia, as well as orotic acid (GC-MS) and homocitrulline were not elevated. She was treated with pyridoxine (500 mg/day) and low protein diet (0.8 g/kg/day) but not significant reduction plasma ornithine level was obtained until now and without effect in terms visual acuity after 12 months of follow-up (Table 1). The clinical diagnosis of the patient was consistent with ornithine-δ-aminotransferase (OAT) deficiency, unresponsive to pyridoxine treatment variant, and gyrate atrophy of the choroid and retina.
Table 1: Plasma amino acid levels at the diagnosis moment, before treatment (sample I) with Pyridoxine treatment, six months after the diagnosis; (sample II) and with pyridoxine treatment and low protein diet; one year after the diagnosis (sample III). View Table 1
The gyrate atrophy of the retina and choroid usually affects individual white. There is only one case described a family African belonging to the black racial group [33]. Also affect both sexes, with onset in childhood. Patients with GA initially complain of decreasing visual acuity and loss of night vision [34]. Eventually, loss of central vision occurs in the fourth to fifth decades [27]. The fundus in patients with GA exhibits circular, well demarcated chorioretinal atrophy with hyperpigmented margins in the midperiphery [35]. Patients with GA may also have myopia and posterior subcapsular cataracts [36]. Usually the fundus finding of scalloped chorioretinal atrophy in the midperiphery is sufficiently characteristic to determine GA. Advanced choroideremia with generalized atrophy of the retinal pigment epithelium and choriocapillaris may be confused. Choroideremia is an X-linked disorder and the macula may be involved earlier than GA. The other differ¬ential diagnosis includes diffuse choriocapillaris atrophy, generalized choroidal dystrophy and central areolar choroidal dystrophy. Plasma ornithine levels help to confirm GA, is a genetic disorder caused by OAT deficiency that results in markedly elevated levels of ornithine in plasma and other body fluids [37]. The exact mechanism of chorioretinal atrophy due to hyperornithinemia is not known, although a low-arginine diet and Pyridoxine supplementation may decrease plasma ornithine levels and reduce the progression of GA [9]. However, the long-term effects of this treatment approach have not been completely evaluated. More than 60 different mutations in the OAT gene locus have been identified [38-40].
Patients with GA generally present with decreased night vision and high myopia during their adolescence, and early diagnosis in these patients allows for early treatment. Indeed, OAT gene studies in children with a high degree of myopia, especially in cases in which there is a complaint of nyctalopia, would be helpful for early identification. To the best of our knowledge, this is the first report of GA in the Cuban population diagnosed by ornithine levels in blood and treated with Pyridoxine dietary supplementation and low protein diet.
Two distinct entities are associated with significant increment in plasma ornithine concentration: Gyrate Atrophy of the choroid and retina (MIM 258870), and the Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) Syndrome (MIM 238970). In patients with gyrate atrophy, visual symptoms first appearance typically occurs at late childhood and after the first few months of life, plasma ornithine concentration ranges from 400 to 1300 µM and plasma ammonium is not elevated. However, the HHH syndrome usually presents with plasma ornithine levels lower than in gyrate atrophy (200 to 1100 µM), while ammonium and glutamine plasma concentrations are increased, particularly after ingestion of a protein load. Urinary excretion of ornithine and its δ-lactam are increased in both types of hyperornithinemia, while homocitrulline usually is not present in urine, is found only in the HHH syndrome. Urinary excretion of orotic acid also is incremented in individuals with HHH [13].
Clinical and biochemical findings for this case suggest the diagnosis of AG; however, molecular studies could not be carried out to identify the underlying mutation.
The patient is the first case diagnosed in Cuba with AG associated with hyperornitinemia. It is a new challenge to perform the molecular study to identify our patients' mutations. The accessibility of genetic services will allow the identification of new cases.
We would like to acknowledge the assistance of Ophthalmologists from Neurophthalmology Department of ICO "Ramón Pando Ferrer", La Habana. Cuba.
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2013. Informed consent was obtained from the guardians of the patient for being included in the study. In addition, the research protocol was reviewed and approved by the medical ethics committee of the National Center of Medical Genetics.
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.