Hurler-Scheie disease is an intermediate form of mucopolysaccharidosis type I. It is a rare metabolic disease that is transmitted in an autosomal recessive mode and causes overload of mucopolysaccharides in all organs including the eye.
Apart from the classically described clinical manifestations, our case is distinguished by the presence of rapidly evolving glaucoma and an easily identifiable retinal surcharge in the fundus as a deposit of whitish chalky substances in the papillomacular bundles.
These findings underscore the importance of regular follow-up of mucopolysaccharidosis patients, including systematic and repeated retinal-photography and even Optical coherence tomography when any abnormality is suspected.
Mucopolysaccharidosis, Retinal overload, Hurler-scheie disease, Glaucoma, Corneal opacity
Hurler-Scheie disease is an intermediate form of mucopolysaccharidosis type I. It is a rare metabolic disease that is transmitted in an autosomal recessive mode and causes overload of mucopolysaccharides in all organs [1] including the eye.
Mucopolysaccharidosis I typically generate a significant hyperopia and astigmatism, corneal opacities, dry eyes, scleral thickening [2], pigmentary retinopathy, papilledema and then optic atrophy. Very rarely, glaucoma can be associated due to the accumulation of glycosaminoglycans within trabecular cells [3].
Our case is distinguished by the presence of rapidly evolving glaucoma and a deposit of whitish chalky substances within the papillomacular bundle of one eye while the papillomacular bundle of the other eye demonstrated striae.
An 8-year-old girl from western Algeria, born in a family with a high rate of consanguinity over many generations presented to the clinic. Like her younger sister, she has Hurler-Scheie disease confirmed by enzymatic activity assay and gene testing that found an enzymatic deficiency of alpha L-iduronidase and IDUA gene mutation. Since the age of seven, she has been receiving enzyme replacement therapy based on recombinant human alpha-L-iduronidase at a dosage of 100 IU/kg/week.
The systemic examination found a particular facies with coarse "gargoyle-like" features, cardiopathy, organomegaly, umbilical hernia, multiple bone deformations secondary to progressive joint stiffness. There is, however, no intellectual disability.
The ophthalmologic examination revealed in both eyes a visual acuity corrected to 5/10, myopia and astigmatism (-2.75 (-2.00) 165° in the right eye and -0.50 (-2.50) 15° in the left eye), ocular dryness, anterior stromal whitish corneal opacities (Figure 1), open-angle chronic glaucoma with an intraocular pressure exceeding 30 mmHg despite well-conducted treatment including dorzolamide, timolol and travoprost, and enlarged optic disc excavation with a cup/disc ratio between 0.8 and 0.9. The examination of the fundus also found areas of the retinal pigment epithelium alteration which can be attributable to atrophy of the retinal pigment epithelium from Myopia (although a bit premature in such a young patient). This may be a precursor to development of pigmentary retinopathy.
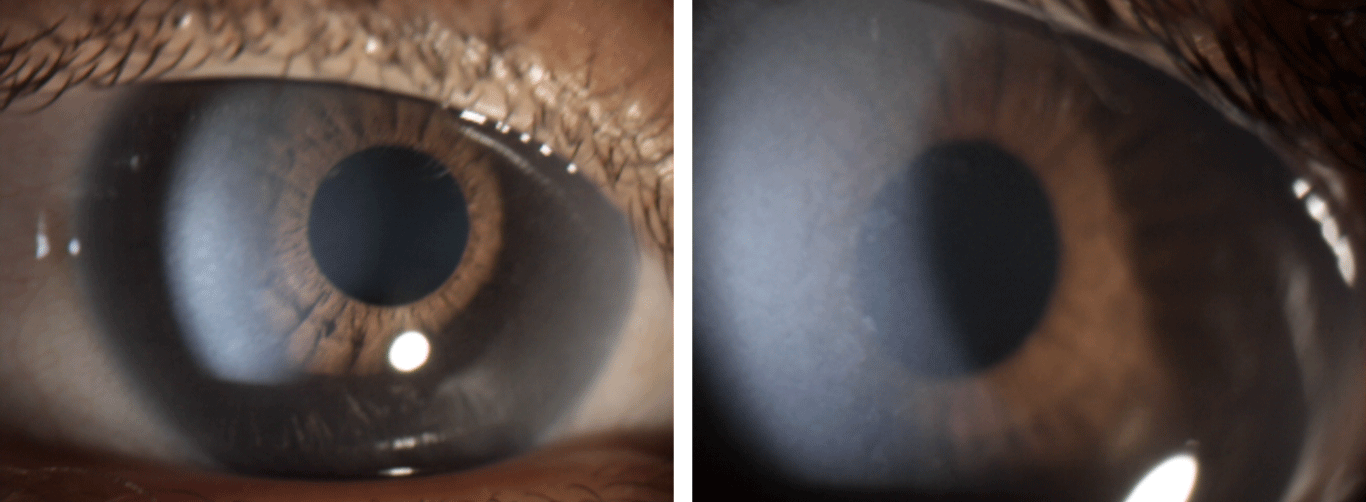 Figure 1: Photographs of the anterior segment objectifying anterior stromal whitish corneal opacities, punctiform and confluent in places.
View Figure 1
Figure 1: Photographs of the anterior segment objectifying anterior stromal whitish corneal opacities, punctiform and confluent in places.
View Figure 1
Chalky white deposits prominent in the papillomacular bundle of the left eye, and retinal striae in the right eye were also found. This is likely related to glycosaminoglycan deposition within the ganglion cells in this region (Figure 2). The macular OCT (optical coherence tomography) finds a thinning of these areas at the expense of the inner retinal layers (Figure 3).
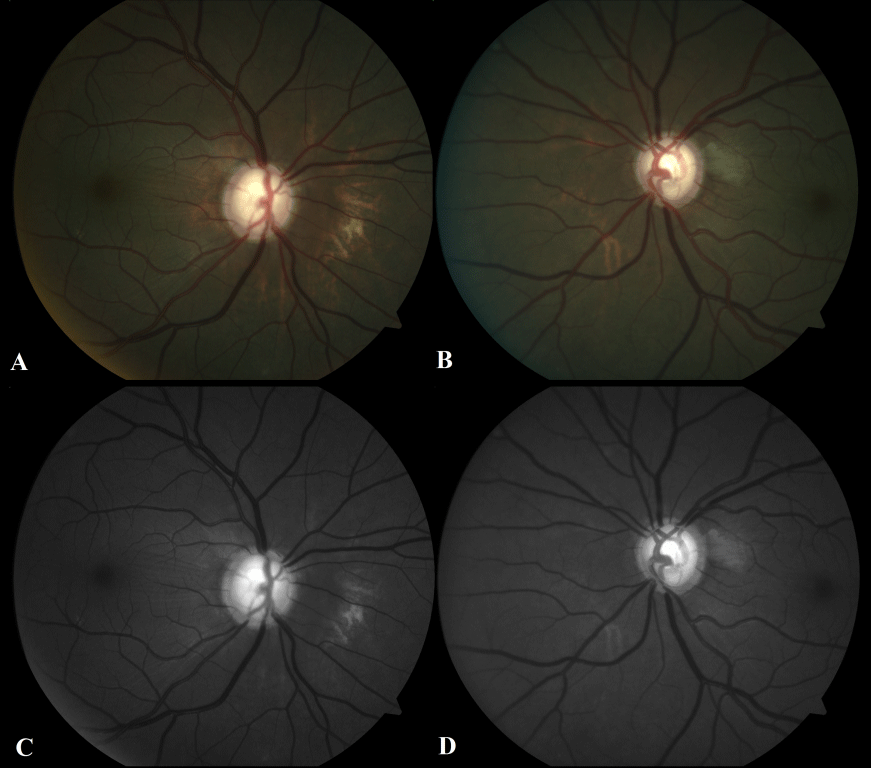 Figure 2: Color retinophotographs (A and B) and with green light (C and D) showing areas of the retinal pigment epithelium alteration and a range of deposits of whitish chalky substances in the papillomacular bundles, better shown on the left eye.
View Figure 2
Figure 2: Color retinophotographs (A and B) and with green light (C and D) showing areas of the retinal pigment epithelium alteration and a range of deposits of whitish chalky substances in the papillomacular bundles, better shown on the left eye.
View Figure 2
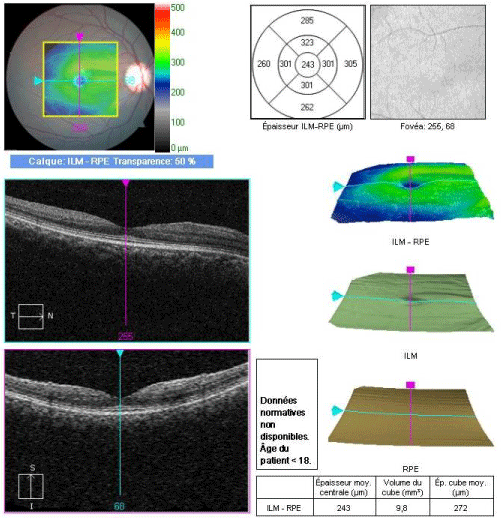 Figure 3: Macular OCT of the right eye showing a normal foveolar profile and a thinning of the papillomacular bundle at the expense of the inner retinal layers.
View Figure 3
Figure 3: Macular OCT of the right eye showing a normal foveolar profile and a thinning of the papillomacular bundle at the expense of the inner retinal layers.
View Figure 3
This retinal involvement not previously described in the medical literature evokes an overload of the inner retina completing the panoply of ocular signs mentioned above.
As for the evolution of this lesion, only regular follow-up and further investigation will enable us to clarify this subject.
All the same, we think that it could possibly evolve towards an atrophy or even a loss of retinal tissue as was the case with another girl with Hurler-Scheie disease, whose macular OCT (optical coherence tomography) found areas of macular thinning with perifovial foci marked by a loss of retinal tissue (breach) at the expense of the inner retinal layers [4].
These findings underscore the importance of regular follow-up of Mucopolysaccharidosis patients, including systematic and repeated retinal-photography and even OCT when any abnormality is suspected [5].