Amyloidosis is a rare disease characterized by deposition of misfolded amyloid protein, which commonly affects myocardium, nerves, kidneys and various organs. The clinical presentation is highly variable. We herein describe, to our knowledge, the first case of a Chinese man with hereditary transthyretin related cardiac amyloidosis (ATTR) with Ala117Ser mutation in Hong Kong who presented with peripheral neuropathy and congestive heart failure. Echocardiography showed left ventricular hypertrophy, with strain analysis demonstrating "cherry on top pattern". Cardiac MRI revealed diffuse subendocardial enhancement on late gadolinium images. There was diffuse tracer activity at heart on Tc99m-pyrophosphate scan. Serum paraprotein was not detected with normal kappa-to-lambda ratio. Genetic tests confirmed a missense mutation Ala117Ser affecting the transthyretin gene. Patient was referred for genetic counseling. New diagnostic strategy and novel treatment are emerging. This case report highlights the importance of low threshold of clinical suspicion to facilitate early diagnosis and hence early treatment.
Transthyretin cardiac amyloidosis, Heart failure, Non-biopsy diagnosis
Hereditary transthyretin related cardiac amyloidosis (ATTR) is a rare disease which is often lethal if left untreated. Previous study in Hong Kong [1] described a case of hereditary transthyretin amyloidosis with Ala117Ser mutation presenting with peripheral neuropathy alone and reported that ATTR amyloidosis was frequently underdiagnosed among Chinese population. Here we report a case of a Chinese male patient with ATTR amyloidosis with cardiac and neurological manifestation.
A 66-year-old Chinese man presented with progressive bilateral lower limb numbness for 6 months in 2015. Patient was an ex-smoker, ex-drinker who had past history of hepatic flexure tumour with right hemicolectomy done. Few months later, patient developed new-onset atrial fibrillation and was hospitalized for congestive heart failure. Trans-thoracic echocardiogram done in another hospital showed LVEF 60-65% with increased left ventricular wall thickness (20 mm). There was mild stenosis (30-40%) in distal left main, ostia to mid left anterior descending artery as well as proximal first diagonal branch on computed tomography coronary angiogram. Nerve conduction test confirmed right carpal tunnel syndrome and axonal sensorimotor neuropathy affecting lower limbs.
Patient was recurrently admitted for congestive heart failure and he remained hypotensive which precluded further titration of heart failure medications. He developed progressive wasting over thenar muscle and small foot muscle. Echocardiogram was repeated in our hospital in 2018, which showed marked increase in wall thickness in both LV and RV. LV strain analysis showed reduced basal and mid longitudinal systolic function with apical sparing ("cherry on top pattern") (Figure 1). Cardiac MRI revealed impaired LVEF 40%, difficult nulling of myocardium and diffuse subendocardial hyperenhancement on late gadolinium images, suggestive of cardiac amyloidosis.
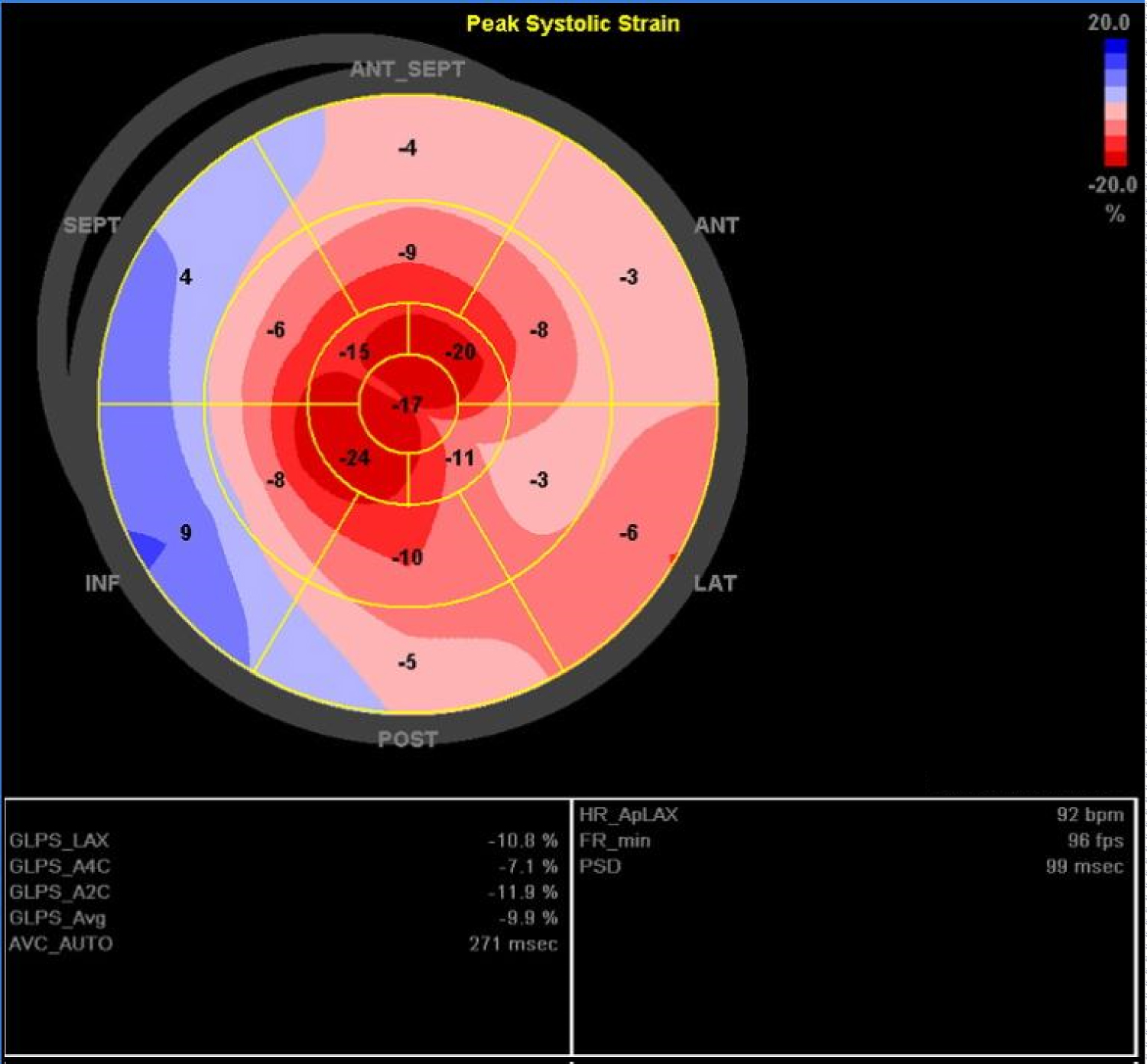 Figure 1: 'Bullseye' display of regional LV longitudinal strain analysis (with mid regions in the middle and apical regions innermost, basal regions outermost).
View Figure 1
Figure 1: 'Bullseye' display of regional LV longitudinal strain analysis (with mid regions in the middle and apical regions innermost, basal regions outermost).
View Figure 1
He had no evidence of plasma cell dyscrasia with negative serum protein electrophoresis/immunofixation for paraprotein and normal serum free light chain ratio. He refused bone marrow biopsy and other biopsy for histological proof for amyloidosis. Subsequent Tc99m-pyrophosphate scan demonstrated diffuse tracer activity at heart, with intensity higher than that of rib (Perugini grade 2 or 3) (Figure 2). Sanger sequencing of all the coding exons and flanking regions of the TTR gene revealed the presence of a heterozygous pathogenic mutation, NM_000371.3(TTR): c.349G > T p.(Ala117Ser) (legacy name: p.Ala97Ser) (Figure 3). Diagnosis of hereditary transthyretin (ATTR) amyloidosis with peripheral neuropathy and cardiomyopathy was established without biopsy.
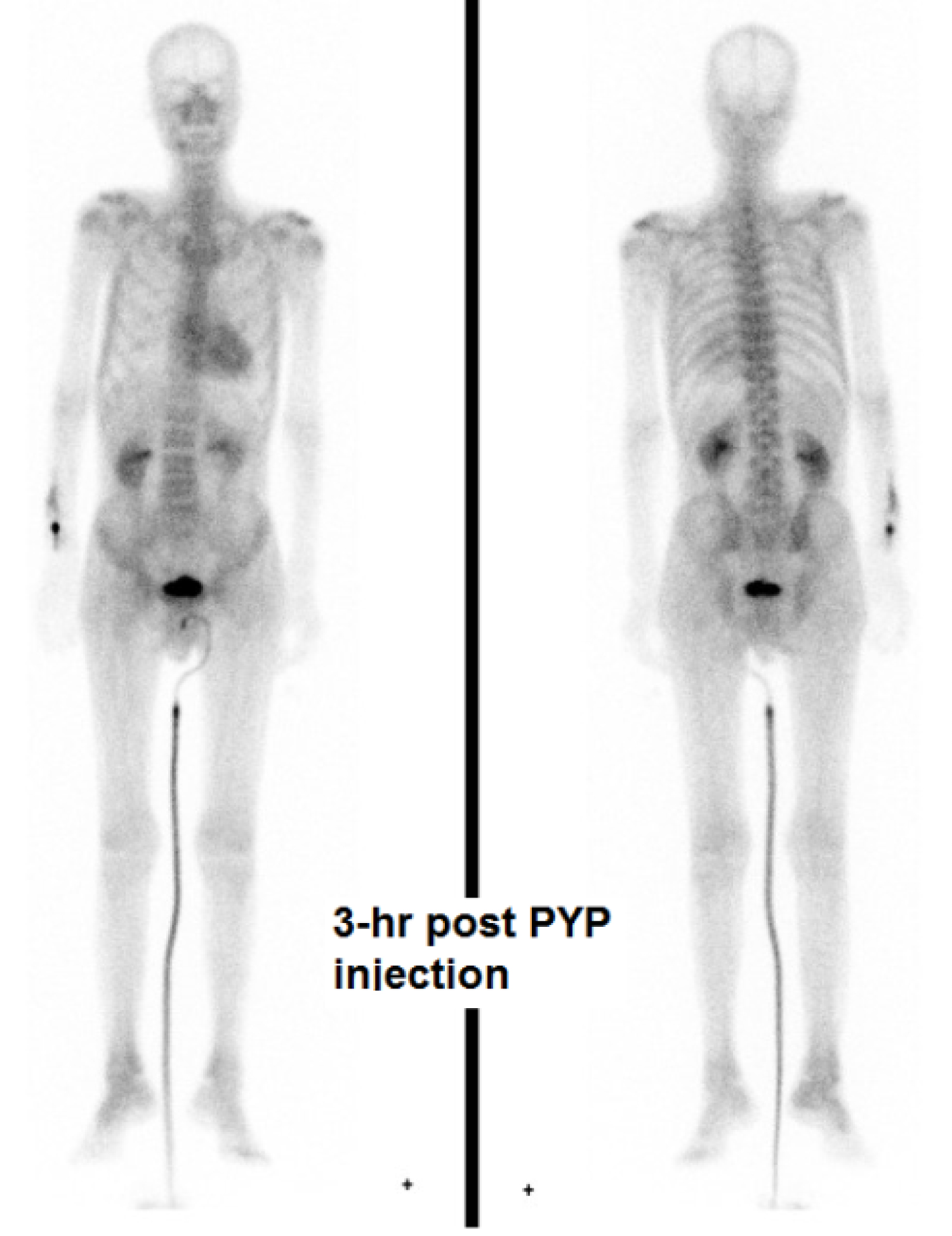 Figure 2: Planar whole body images of Tc99m-pyrophosphate (PYP) scan - 180 min following injection demonstrated diffuse tracer activity at heart.
View Figure 2
Figure 2: Planar whole body images of Tc99m-pyrophosphate (PYP) scan - 180 min following injection demonstrated diffuse tracer activity at heart.
View Figure 2
 Figure 3: DNA sequencing chromatogram of the c.349G > T p.(Ala117Ser) mutation.
View Figure 3
Figure 3: DNA sequencing chromatogram of the c.349G > T p.(Ala117Ser) mutation.
View Figure 3
Currently TTR stabilizer is licensed in Hong Kong only for stage I amyloidosis while diflunisal was not considered in view of the advanced heart failure. Oral treatment with tauroursodeoxycholic acid (TUDCA) 250 mg tds and doxycycline 100 mg bd for polyneuropathy was introduced. However TUDCA was soon discontinued due to marked diarrhoea.
Amyloidosis is characterized by accumulation of misfolded precursor protein, which has undergone conformational changes into beta-pleated sheet configuration. Deposition of the amyloid fibrils in tissues leads to organ damage. Amyloidosis can be categorized according to extent of amyloid deposition (systemic vs. localized) or types of amyloid precursor protein, for example, immunoglobulin light chain, serum amyloid A, transthyretin, which resulted in AL amyloidosis, AA amyloidosis and ATTR amyloidosis respectively. ATTR amyloidosis is further classified into wild type (also known as the senile type, with wildtype DNA sequence in the TTR gene) and hereditary type. Hereditary ATTR amyloidosis, also known as familial amyloid polyneuropathy, is an autosomal dominant disorder with variable penetrance. More than 120 TTR mutations have been reported in different ethnic groups, but infrequently among the Chinese population. A Taiwanese cohort involving 79 hereditary ATTR amyloidosis patients shows that Ala117Ser is the commonest mutation in ethnic Chinese, accounting for 91.2% of ATTR pedigrees. The Ala117Ser genotype is associated with peripheral neuropathy as the first symptom in 80.8% of patients, which occurred in this case [2]. From the exome sequencing data in Genome Aggregation Database (gnomAD), the variant was only observed once in the "Other East Asian" (non-Japanese, non-Korean) normal population with an allele frequency of 0.006934% (one allele found in 14,422 alleles). It was not otherwise observed in the 236,590 alleles in other populations) [3]. While the exact penetrance of in carriers of this variant is unknown, the carrier rate of this mutation is estimated to be 1 in 7,200 in the non-Japanese, non-Korean East Asian population. The clinical manifestations vary with different TTR genotypes, though the majority of patients present with cardiomyopathy or peripheral neuropathy. Diagnosis is frequently delayed as a result of the rarity of the disease and the non-specific clinical presentation.
Echocardiogram is often used as the initial investigation for heart failure. The cardinal echocardiographic features of cardiac amyloidosis include increase in left ventricular wall thickness, diastolic dysfunction and granular sparkling of myocardium. 2D speckle tracking strain analysis measures the degree of myocardial deformation, which is defined as ratio of change in length of myocardial segment relative to its baseline. Myocardial strain can be assessed in three dimensions i.e. longitudinal, radial and circumferential strain. Longitudinal strain refers to myocardial deformation from base to apex in the long axis while radial strain describes the myocardial wall thickening along its radius. Circumferential strain denotes rotational motion as viewed from the apex. Typically, cardiac amyloidosis is associated with impaired basal but preserved apical longitudinal strain. Moreover, in a study of patients with preserved LVEF and mild hypertrophy, an ejection fraction to global longitudinal strain ratio > 4.1 was shown to be 89.7% sensitive and 91.7% specific in differentiating cardiac amyloidosis from other causes of LV hypertrophy [4].
The gold standard of diagnosis of amyloidosis requires tissue biopsy with the demonstration of apple-green birefringence with Congo red staining under polarized light microscopy. Subsequent typing of the amyloid precursor protein is achieved by immunostaining or more recently by laser microdissection and mass spectrometry on tissue samples. However, tissue biopsy is often invasive and associated with long waiting time and hence possible delay in diagnosis. Meanwhile conventional echocardiography is neither sensitive nor specific. Fortunately, there is upcoming evidence regarding non-invasive diagnostic approach in ATTR cardiac amyloidosis. Radionuclide bone scintigraphy using 99mTc-labeled 3,3-diphosphono-1,2-propanodicarboxylic acid (DPD) or 99mTc-labeled pyrophosphate (PYP) as tracer can identify early cardiac ATTR deposition and aid the differentiation from other types of cardiomyopathy. Uptake of DPD or PYP in the heart equivalent to or greater than that in the bone (Perugini grade 2 or 3), combined with the absence of serum and urine monoclonal protein is diagnostic of ATTR amyloidosis with 100% positive predictive value [5]. Consequently, the diagnosis of hereditary ATTR amyloidosis is established in our case by this non-invasive criterion, along with the positive genetic test. The improvement of imaging techniques, namely the availability of cardiac MRI and radionuclide scintigraphy, was found to be associated with an increase in referrals for TTR genetic analysis, leading to an increase in diagnoses of both wildtype and hereditary transthyretin amyloidosis [6]. Genetic diagnosis of hereditary transthyretin amyloidosis by Sanger sequencing involves sequencing of four exons and is helpful for the disease confirmation, family screening with identification of presymptomatic carriers as well as exclusion of wildtype transthyretin amyloidosis.
TTR plasma protein is primarily produced by the liver. Therefore, liver transplantation has historically been the standard of care for hereditary ATTR amyloidosis to replace the source of mutant TTR protein. In 2018, three phase 3 trials using TTR stabilizer (Tafamidis [7]) and TTR RNA interference agents (Patisiran [8] and Inotersen [9]) demonstrated slower rate of functional decline and improved clinical outcome when comparing with placebo among patients with ATTR amyloidosis. The availability of new therapeutic options highlights the importance of early diagnosis, family screening and potentially regular monitoring in asymptomatic mutant TTR carrier.
Here we describe a Chinese man with hereditary ATTR cardiac amyloidosis with peripheral neuropathy in Hong Kong. New diagnostic strategy and novel treatment are available. This case report illustrates the significance of low threshold of clinical suspicion to facilitate timely diagnosis and thus early treatment.
The authors declare that there is no conflict of interest.