A 54-year-old male with a history of Maffucci syndrome and Marginal Zone Lymphoma, presented with a 4-week history of headache, right-sidedptosis and diplopia. Whole-body imaging revealed a mass in the pituitary fossa that was likely lymphomatous. Despite diagnostic uncertainty, the mass was treated as transformation of Marginal Zone Lymphoma to a high-grade lymphoma. This report analyses how and why the multi-disciplinary team treated the patient without a biopsy. The case highlights a possible novel association between Maffucci syndrome and either a high-grade lymphoma or Marginal Zone Lymphoma. A genetic explanation for this combination of rare disease may exist due to a shared association with a mutation at 14q11.2.
Maffucci syndrome is a rare, non-hereditary disease, characterized by the sporadic occurrence of multiple enchondromas combined with cutaneous, visceral and/or soft tissue hemangiomas [1,2]. A study from 2008 reported fewer than 200 documented cases of Maffucci Syndrome worldwide [3]. Somatic mutations in Isocitrate dehydrogenase 1 (IDH1) or IDH2 genes are common amongst those with Maffucci syndrome and these mutations have oncogenic potential [4]. Furthermore, patients with Maffucci syndrome have other oncogenic mutations at the regions 2p22.3, 2q24.3 and 14q11.2 [5]. These mutations cause epigenetic modifications which can cause changes in the way deoxyribonucleic acid acid (DNA) is expressed which activate cancer related genes or tumours. Hemangiomas and enchondromas have a risk of transforming into vascular sarcomas and chondrosarcomas respectively [6]. Due to the rarity of Maffucci syndrome, rarer malignant associations of the disease are unlikely to be documented.
Marginal Zone Lymphoma (MZL) is a sub-set of indolent Non-Hodgkin B-cell Lymphoma (NHL). MZL is thought to arise from memory B lymphocytes in the marginal zone of lymphoid tissue [7]. There are three subtypes of MZL and they differ on site predilection. The three types are splenic, nodal and mucosa-associated lymphoid tissue [8]. Nodal Marginal Zone Lymphoma and Splenic Marginal Zone Lymphoma typically present between 50 and 60 years of age. They affect males and females equally and patients normally present with disseminated disease, but central nervous system (CNS) involvement is uncommon [9-11]. The 5 year survival rate of nodal MZL, in the current Rituximab-treated era, has reached 70-90% [12]. The MZL transforms to Diffuse Large B- Cell Lymphoma in 15% of patients [13]. There is no standard of care for the treatment for MZL but options include surgery and/or radiotherapy for truly localised disease (rare) and immune-chemotherapy for more disseminated disease. Patients who are asymptomatic and have no bone marrow involvement will usually be managed with close observation/watch and wait approach [14].
The author is not aware, to date, of documented cases of MZL or High-grade Lymphoma in a patient with Maffucci Syndrome. However, there are mutations that can be present in Maffucci Syndrome (2q24.3 and 14q11.2) that are present in Lymphomas. Notably mutation 14q11.2 has been associated with Maffucci syndrome and Burkitt's Lymphoma meaning that theoretically Maffucci syndrome may be associated with high-grade lymphoma [5] (Table 1).
Table 1: Mutations associated with Maffucci Syndrome and Haematological malignancy ref (common somatic alterations identified in maffucci syndrome by molecular karyotyping) [5]. View Table 1
In February 2018 a 54-year-old male presented to the emergency department with a 4 week history of progressive persistent right-sided frontal headache and progressively deteriorating blurred vision and right-sided ptosis. The patient had a past medical history of Maffucci syndrome, MZL and Hyperlipidaemia. On examination he had a right-sided ptosis, dilated pupil and ophthalmoplegia consistent with a 3rd and 4th nerve palsy. There was pain over the ophthalmic division of the 5th cranial nerve consistent with a unifying clinical diagnosis of cavernous sinus syndrome. All other cranial nerves were intact and the peripheral nervous examination was normal. There were no systemic manifestations on examination to suggest hypo- or hyperpituitarism.
Maffucci syndrome was diagnosed in 2006 following investigations for pain in the right hand. Both enchondromas and a haemangioma, were found, both of which are known to be diagnostic of Maffucci syndrome [2]. The patient also had a history of MZL. MZL was diagnosed from an inguinal lymph node biopsy in September 2017 following a prior presentation with inguinal lymphadenopathy, mild thrombocytopenia and splenomegaly. CT Chest Abdomen Pelvis (CAP) at this time also showed widespread involvement including supra- and infra- diaphragmatic nodes as well as splenomegaly. MZL was being managed with expectant observation only as thrombocytopenia resolved, bone marrow biopsy had confirmed that there was no bone marrow involvement and there were no related symptoms.
CT head showed no obvious intracranial findings in the brain or pituitary fossa. Lumbar puncture showed raised cerebrospinal fluid (CSF) protein (0.6 g/L) with normal glucose (3.5 mmol/L); no organisms were seen on CSF microscopy or gram staining. Mature lymphocytes were present in the CSF with no evidence of clonality on immuno phenotyping. CSF analysis by PCR excluded Epstein Barr Virus, Cytomegalovirus, Human Herpes virus 6 and Human Herpes virus 8. Magnetic Resonance Imaging (MRI) head with contrast showed a 13 × 37 × 13 mm homogeneously-enhancing sellar mass which invaded the right cavernous sinus with mild displacement of the optic chiasm (Figure 1a). Pituitary function was normal. A CT CAP had shown the widespread lymphadenopathy was stable in size compared with previous imaging from initial diagnosis of MZL. A staging whole body fluorodeoxyglucose (FDG)-positron emission tomography (PET)CT scan showed an FDG-avid lesion with a maximum standardized uptake value (SUVmax) of 11.6 g/ml in the cavernous sinus together with widespread supra- and infra-diaphragmatic FDG-avid nodal disease with splenic involvement.
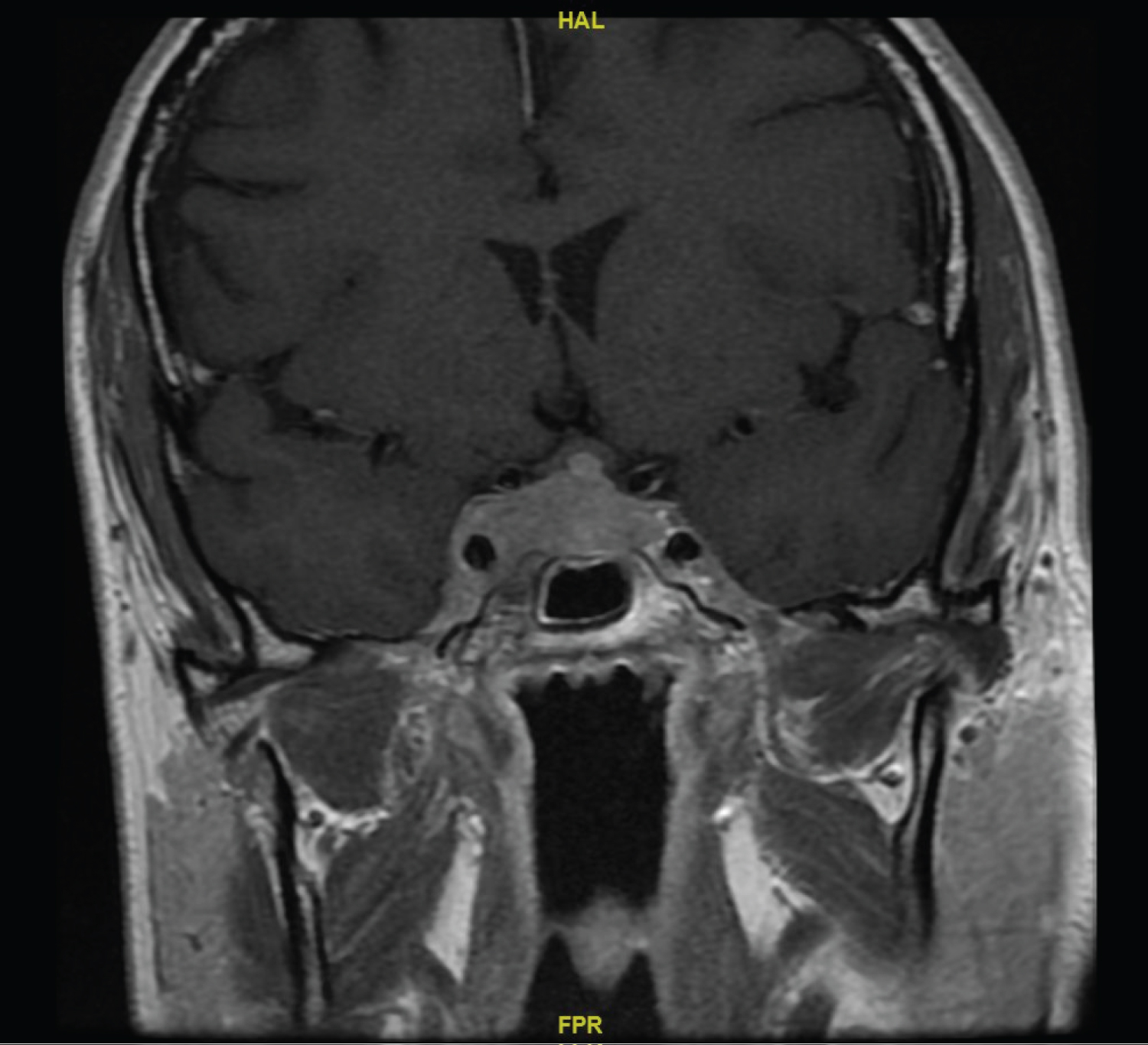 Figure 1a: MRI showing right-sided cavernous sinus invasion at presentation.
View Figure 1a
Figure 1a: MRI showing right-sided cavernous sinus invasion at presentation.
View Figure 1a
The lesion could have represented either a transformation of the MZL to a high-grade lymphoma, infiltration of the benign MZL into the CNS or could represent other primary pituitary lesions as discussed later. The hematology team discussed the case with Endocrinology and Neurosurgery and made the decision to treat the lesion as high-grade lymphoma and forego a biopsy in view of associated risks and the patient's rapidly evolving neurology. The patient was given empirical treatment with high-dose intensive CNS-penetrating chemotherapy consisting of rituximab, vincristine, doxorubicin, cyclophosphamide, methotrexate, etoposide, ifosfamide, cytarabine and steroids (R CODOX M IVAC). This represents an intensive inpatient regime with high morbidity including acute infection risk, hepatotoxicity, nephrotoxicity and risk of secondary malignancy [15,16]. The main complications through the 4 cycles of chemotherapy were an axillary abscess, an episode of neutropenic sepsis and acute renal injury (nadir estimated glomerular filtration rate (eGFR) was 13 ml/min/1.73 m2). These complications were all treated and subsequently resolved after treatment.
MRI head 6 months after chemotherapy showed a significant reduction in the size of the mass in the pituitary fossa which no longer compressed cranial nerves. Cranial nerve palsies had also resolved. Before the most recent review, MRI head 21 months following chemotherapy showed resolution of the sellar mass (Figure 1b). At this time CT CAP showed complete resolution of supra- and infra- diaphragmatic lymphadenopathy. Furthermore, full blood count values were within reference ranges and endocrine status was grossly normal. This suggested disease remission.
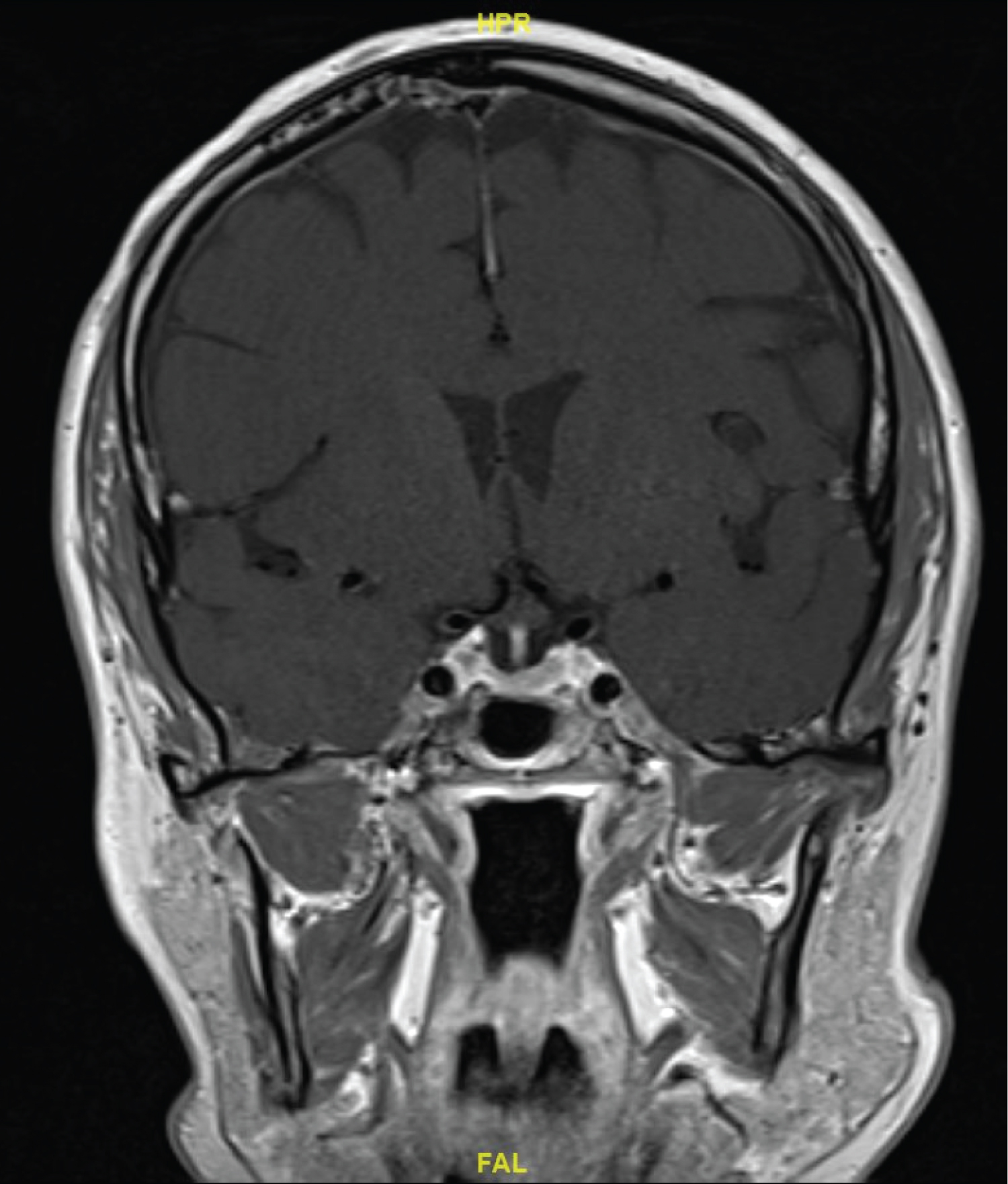 Figure 1b: Resolution of cavernous sinus mass after chemotherapy.
View Figure 1b
Figure 1b: Resolution of cavernous sinus mass after chemotherapy.
View Figure 1b
This case is important because it raises a potential novel association between Maffucci syndrome and either a high-grade transformation of MZL or a rapidly growing MZL at an unusual site. The case also raises the clinical conundrum of the underlying diagnosis of the pituitary lesion. Theoretically the pituitary lesion could have represented a high-grade transformation of MZL, a rapidly-growing CNS infiltration of MZL, a vascular or inflammatory process or a primary tumour. The differentials of a primary tumour are wide as Maffucci's syndrome has been associated with multiple intracranial tumours including pituitary adenomas, astrocytomas, olfactory neuroblastomas, malignant chordomas and spindle cell hemangioendotheliomas [17-20].
In view of the unknown nature of the pituitary lesion consideration was given to obtaining a biopsy. Some groups have advocated gaining a histological diagnosis in similar clinical scenarious previously whilst others have taken a more conservative approach because of the potential neurovascular complications of taking a biopsy of a lesion in the cavernous sinus [21]. Furthermore, consideration was given to biopsy a peripheral node with similar SUV uptake however the speed at which the patient's neurology was evolving meant a decision was made on clinical grounds to treat the patient.
In view of how rare CNS involvement from MZL is, the speed at which the patient's neurology was appearing, the SUV level of the lesion, and the spread of lymph node involvement, transformation of MZL to a high-grade lymphoma was thought to be the most likely diagnosis. It would have been very unlikely for MZL to present with such rapidly evolving symptoms. Furthermore, the similar SUV level across multiple lesions made the pituitary lesion unlikely to have a distinct primary origin from the other lesions. As mentioned, the SUVmax of 11.6 g/ml was significant in reaching a diagnosis as groups comparing the SUVmax levels in aggressive and indolent Non-Hodgkin Lymphoma found that an SUVmax of 10 g/mL and above predicts aggressive lymphomas with high specificity and sensitivity [22].
In view of these reasons to support the likelihood that the cavernous sinus mass represented a high-grade lymphoma and the risks of obtaining a biopsy from the pituitary mass, the MDT decided that a biopsy was not in the patient's best interests.
Fortunately, a good response to empirical treatment with chemotherapy was seen with apparent disease remission suggesting that the urgent and pragmatic decision to treat a high-grade lymphoma was a good decision. Furthermore this response reaffirmed the working diagnosis of either high-grade or low-grade lymphoma as an alternative primary CNS tumour was unlikely to respond as favourably given the poor prognosis of primary CNS tumours such as astrocytoma (median survival of 14.6 months and 2-year survival rate of 26% [23,24].
This case presents a difficult scenario where the team was dealing with uncertainty. The decision not to biopsy the cavernous sinus lesion nor the axillary node meant the management plan was based on an unknown diagnosis. It shows the value of a logical MDT decision and that if thought through, maximal investigation is not always in the patient's best interests.
For reasons described, this report likely demonstrates a novel case of a patient with both Maffucci syndrome and high-grade transformation of MZL. An association between Maffucci Syndrome and Lymphoma may be explained by a genetic mechanism involving a mutation at 14q11.2 [5]. Had a biopsy been taken, it would have been interesting to look at Whole Gene Sequencing (WGS) of the tumour to investigate if a more comprehensive unifying genomic explanation exists. With the advent of increasingly available WGS within mainstream genetic testing, clinical cases such as this will be increasingly useful in determining the molecular explanations for rare disease combinations such as described in this case [25].
This case demonstrates how a logical MDT discussion can result in speedy and likely appropriate treatment of an unknown tumour. It also highlights a novel association between Maffuci syndrome and either high-grade transformation of MZL or a rapidly growing low-grade lymphoma in an unusual site. More observational studies should be performed involving patients with Maffucci syndrome to examine its potential association with MZL, in particular high-grade transformations of MZL. In view of the limited evidence base, clinicians should be aware that an association may exist and continue to report further associations.