The human skeleton is composed of bone, a living tissue that undergoes constant development throughout life. It is well established that changes in bone metabolism during the developmental stages of growth, modelling and remodelling determine long-lasting physiological parameters, such as final height achieved, peak bone mass, bone quality and bone health. A complex interplay of environmental, genetic, nutritional, physiological and behavioural factors plays a role in these processes. These modifiable and non-modifiable factors influence skeletal development and bone quality, as well as the occurrence of clinical conditions during adulthood, such as osteoarthritis and osteoporosis.
The phase of skeletal growth is a critical period in childhood development. Factors that adversely impact bone growth in childhood will have lifelong consequences, including short stature in adulthood and potentially also deficits in bone geometry, bone strength and bone structure.
X-linked hypophosphataemia (XLH) is an example of a genetic condition that exerts a significant negative impact on skeletal growth and development. This rare inheritable disease is characterized by chronic renal phosphate wasting, where the lack of phosphate during the pivotal period of bone development especially in periods of rapid growth leads to rickets and poor longitudinal growth, resulting in limb deformities and disproportionate short stature.
Understanding skeletal development in disorders associated with impaired growth is important to accurately identify growth patterns that deviate from the expected norm. The absence of a universally accepted definition for a growing skeleton poses a challenge for assessing end-of-growth in young adults. The authors reviewed key aspects of skeletal development, how skeletal growth is currently defined and measured, and propose definitions of a growing and maturing skeleton that would be applicable in the clinical setting, both in health and in pathological growth disorders, such as XLH.
Skeletal growth, Growing skeleton, Maturation, Final height, X-linked hypophosphataemia
XLH: X-Linked Hypophosphatemia; IGF-I: Insulin-like Growth Factor 1; SH: Sitting Height; PBM: Peak Bone Mass; BMC: Bone Mineral Content; aBMD: areal Bone Mineral Density; vBMD: volumetric Bone Mineral Density; DXA: Dual X-Ray Absorptiometry; HR-pQCT: High-Resolution Peripheral Quantitative Computed Tomography; CT: Computed Tomography; 1,25(OH)2D: 1,25 Dihydroxyvitamin D; PTH : Parathyroid Hormone; PHEX: Phosphate-Regulating Endopeptidase Homolog X-Linked; FGF23: Fibroblast Growth Factor 23; MAPK : Mitogen-Activated Protein Kinase; BDAT: Bidirectional Axial Transmission; MRI: Magnetic Resonance Imaging; TW2: Tanner-Whitehouse 2nd Edition; SDS: Standard Deviation Score; ASL: Arm Span Length; ASHR: Arm Span to Height Ratio; IC : Intercondylar; IM: Intermalleolar; TBMC: Total Body BMC; TBMD: Total Body BMD
The human skeleton serves multiple essential functions that include providing support for the rest of the body; providing levers for the muscles to allow movement and locomotion; protecting vital internal organs and structures; providing maintenance of mineral homeostasis and the acid-base balance; serving as a reservoir of growth factors and cytokines; and providing an environment for haematopoiesis within the marrow spaces [1]. The skeleton undergoes major size changes and adaptations to forces, during infancy and childhood. Imbalances in nutritional status, diseases of many origins or constitutional/genetic conditions, may affect skeletal growth and maturation, often leading to shorter stature and/or altered bone structure/geometry.
Measuring skeletal growth (as height) and body proportions as well as evaluating skeletal radiographs are useful for identifying and diagnosing children with short stature or growth disorders; the projected adult height of children is compared with the 'target height' or genetic height based on parental height [2].
X-linked hypophosphataemia (XLH), is a rare lifelong hereditary disorder characterized by renal phosphate wasting leading to chronic hypophosphataemia [3]. The altered phosphate metabolism produces skeletal mineralization abnormalities with growth retardation and short, disproportionate stature being major manifestations of the condition. Since bone undergoes growth, modelling and remodelling processes during childhood; remodelling and, to a lesser extent, modelling in adult life [4,5]; an adequate supply of essential elements, including calcium and inorganic phosphate, is critical to maintain a healthy skeletal structure throughout life.
In disorders of growth or bone pathology, such as XLH, there is a need to define the end-of-skeletal growth. The current lack of a universal definition for the 'growing skeleton' poses a challenge to the optimal management of disorders with impaired skeletal growth and development since the aptness of certain treatments is dependent on whether or not longitudinal growth has ceased. Similarly, the definition of the attainment of final height is also inconsistent.
The authors of this paper held several working sessions to develop a standard definition for the growing skeleton that would be applicable in health and diseases with pathologic skeletal growth and development. Suggestions for the definition of skeletal growth and maturation were: (1) 'a growing skeleton increases in height and shows radiological non-fusion of growth zones' and (2) 'in children and adolescents, a skeleton should be considered to undergo skeletal maturation as long as it is accruing bone mass and bone density'.
This review article briefly describes the physiology and key milestones of normal skeletal development. Skeletal deficits observed in conditions that are associated with pathologic growth, such as XLH, are also discussed. Furthermore, the concept of end-of-skeletal growth is discussed in the context of a lack of a universal definition. An updated definition for a 'growing and maturing skeleton' is proposed.
There are four categories of bones that make up the human skeleton: long bones, short bones, flat bones and irregular bones. These different bone types undergo one of two distinct processes of skeletal development depending on the embryological origin, namely, intramembranous or endochondral ossification [6,7].
Intramembranous ossification is the process of replacing sheet-like connective tissue with bony tissue. This process produces most of the flat bones, i.e., craniofacial bones and part of the clavicles. Bone development occurs from mesenchymal condensations, differentiating directly into osteoblasts in the primary ossification centres without any prior cartilage formation [6-10].
Increases in human height are primarily accounted for by endochondral ossification, the process responsible for development of the long bones. In the embryological phase, chondrocytes initially create a cartilage model, followed by the invasion of osteoblasts and blood vessels into the skeletal element to create the primary ossification centre in the middle of the bone. This forms a denser bone matrix known as primary spongiosa [11]. The cartilage matrix continues to form as the foetus grows, lengthening the bone, with layers of rounded 'resting' chondrocytes at the ends and flat 'proliferative' chondrocytes towards the middle. Postnatally, secondary ossification starts to occur at centres at the end of the long bones. The growth plate can be found in between the primary and secondary ossification centres [11]. Any disturbance of the plate physiology may cause developmental abnormalities [6,10,12,13].
Longitudinal growth is driven by elongation of the bones caused by the proliferation and differentiation of chondrocytes at the epiphyseal growth plates [6,14,15]. The growth plate is a structure present in growing children, composed of columns of chondrocytes found in a germ layer near the epiphyseal bone. Chondrocytes grow in size until they are replaced by bone in the metaphyseal bone, following apoptosis and vessel invasion [11].
The main factor influencing longitudinal growth is the size of the hypertrophic chondrocyte prior to its replacement by bone. Up to 73% of bone lengthening has been attributed to the volume increase achieved by these hypertrophic chondrocytes [16].The regulation of chondrocytes and longitudinal growth is thought to involve both systemic factors (e.g., growth hormone, parathyroid hormone, vitamin D, thyroid hormones, sex hormones, glucocorticoids) and local factors (e.g., insulin-like growth factor 1 (IGF-I), vascular endothelial growth factor, transforming growth factor beta 2, parathyroid hormone-related protein, leptin, integrins, prostaglandins and chondromodulin) [16,17].
A considerable part of longitudinal growth occurs at the vertebral bodies, where the increase in sitting height (SH) accounts for more than half of the height gain during the pubertal growth spurt [18]. The mechanisms of growth in vertebral height are similar to those in long bones, with endochondral ossification taking place in the growth plates adjacent to the discs. Sustained mechanical loading can also modulate growth in height [19]. Vertebral bodies also grow via primary and secondary ossification centres in a process where the vertebrae form shapes that optimize the protection of the spinal cord, aid general mobility and provide support for the head and neck. The vertebral bodies continue changing and adapting via endochondral ossification into adulthood as greater mobility and stability is required [20].
Increases in longitudinal growth of the long bones only occur during the growth phase of bone development. Normal childhood growth is characterized by: (1) a rapid, decelerating infantile growth phase lasting until about 3 years of age; (2) a longer childhood phase with a steady decelerating growth velocity; and (3) the adolescent growth spurt that is marked by an initial period of rapidly accelerating growth velocity followed by deceleration until final adult height is reached [21].
Appositional growth occurs at the diaphysis of the long bone and the vertebral bodies, which changes the bone shape and increases its width [19,22,23]. During appositional growth of the long bones, width is added on the outside through the process of subperiosteal apposition. Layers are added to those that already exist, while bone is simultaneously removed on the inside of the medullary bone cavity via endosteal resorption - the breaking down and reabsorption of bone material from the centre. Appositional growth (bone modelling) conserves the tubulation of long bones when the metaphyseal width is reduced into the diaphyseal dimension; failure in modelling may cause under- or over-tubulation. This continues throughout life, even after the cessation of longitudinal growth, in response to stresses from muscle activity or weight [5,7,24,25].
Bone modelling primarily takes place in actively growing bone, where bones are shaped or reshaped by either bone formation or bone resorption occurring at a given bone surface [4,7,26]. In bone modelling, bone adapts to its function in response to stress from muscle activity or weight by the deposition of bone and removal of bone where required. This process provides increased strength to the bone structure, while minimizing density to maintain its lightness [4,26,27]. Bone modelling can still occur, to a much lesser extent, in the adult skeleton as bones adapt to permanent strain, pressure or fractures [4,5].
Bone remodelling is a process that occurs once new bone has formed. Mineralized bone is removed by osteoclasts; the osteoblasts then replace the bone matrix, which has been resorbed. Osteoclasts and osteoblasts work sequentially in the bone forming unit during this process. Osteoclasts are activated on the bone surface and resorb the mineralized bone and the extracellular matrix. This creates a defect that is filled by the osteoblasts, resulting in new bone formation (Figure 1) [1,4,28-30].
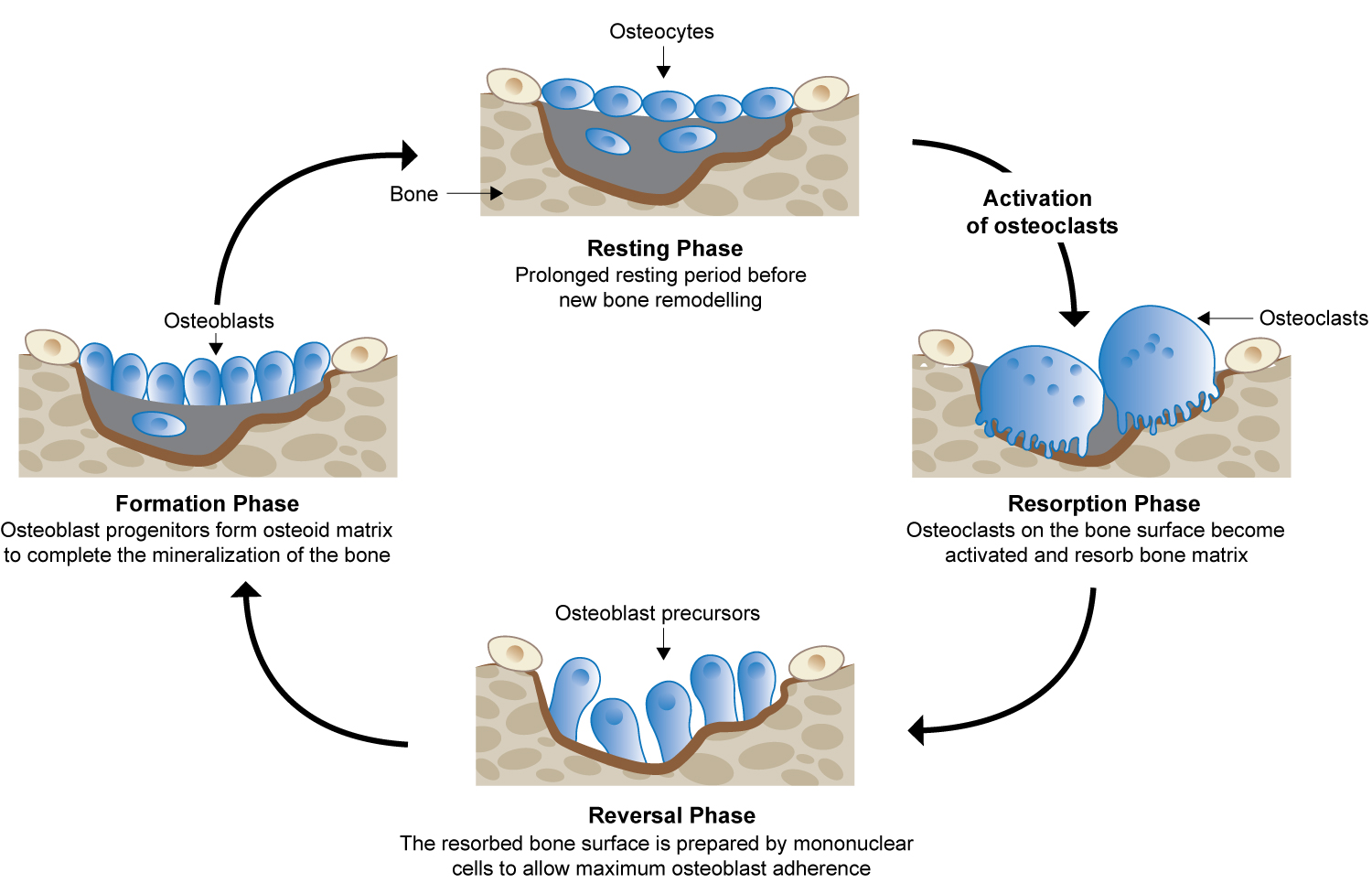 Figure 1: Bone remodelling cycle. Adapted from Moreira, et al. [30].
View Figure 1
Figure 1: Bone remodelling cycle. Adapted from Moreira, et al. [30].
View Figure 1
In childhood, bone remodelling is characterized by a positive bone turnover, with an overall increase in the amount of new bone formed compared with the levels of bone resorption. Once peak bone mass (PBM) is achieved, there is a net balance between bone resorption and bone formation, which then declines with age-related bone degeneration leading to a net loss of new bone. The bone remodelling process repairs microdamage, maintains biomechanical strength and ensures mineral homeostasis (phosphate and calcium) [4,31].
PBM is defined as the maximum amount of bone tissue accrued during an individual's life; this occurs after normal growth has ceased and indicates that the skeleton has fully matured [32-35]. Bone mass is commonly measured by bone mineral content (BMC) and areal or volumetric bone mineral density (aBMD or vBMD) using dual X-ray absorptiometry (DXA) or high-resolution peripheral quantitative computed tomography (HR-pQCT)/computed tomography (CT) [36]. Lifelong bone health is dependent on maximizing PBM during the critical periods of growth, bone mineralization and maturation [37]. The growth years during childhood and young adulthood are particularly important for building PBM. Almost half of adult bone mass is attained during adolescence, with 25% being acquired during and just after the peak height gain during the pubertal growth spurt [38]. A higher risk in later life for fragility fractures appears if optimal PBM is not reached and maintained during adulthood [34,36,39].
In the healthy population, genetics can determine around 60-80% of PBM [35,38,40]. However, the predetermined genetic trajectory for the accrual of PBM can be influenced by modifiable environmental factors, in particular, physical activity and optimal nutritional intake of protein, calcium and vitamin D [33,37,38,40].
The impact of physical activity on bone mass acquisition during childhood is modulated by diet, particularly through the intake of protein and other essential nutrients [33]. Dietary protein enhances the production of IGF-I that stimulates the kidney production of 1,25 dihydroxyvitamin D (1,25(OH)2D), which, in turn, boosts intestinal absorption of calcium and inorganic phosphate, thereby positively influencing bone mineralization [33,41]. This production of 1,25(OH)2D is tightly controlled and is stimulated by parathyroid hormone (PTH). Phosphate (in the form of inorganic phosphate) plays a vital role in bone health and constitutes a major component of bone [42]. Approximately 85% of total adult body phosphate resides in the bone [43]. Adequate phosphate levels are crucial for the apoptosis of hypertrophic chondrocytes and, along with calcium, for mineralization of the newly formed bone matrix. Although dietary intake of phosphate usually exceeds the quantity of phosphate required for these processes, increased excretion of phosphate in patients with XLH results in phosphate deficiency. Maintenance of normal phosphate homeostasis is crucial for normal bone growth, especially during the phases of rapid growth [44].
Up to 50% by volume and 70% by weight of human bone is a modified form of hydroxyapatite, known as bone mineral. It is composed of calcium and phosphate Ca10(PO4)6(OH)2. Since adequate levels of vitamin D are required for the intestinal absorption of calcium and phosphate, [45] an imbalance in vitamin D levels will negatively impact the relationship between dietary calcium and phosphate intake and bone mineral density [38,41,46]. Decreased vitamin D levels have been associated with a low bone mineral content and density, osteomalacia in adults and rickets in children [47]. The nutritional vitamin D level is one of the physiological determinants of PBM, and supplementation in infancy is associated with increased aBMD [40,48]. Murine models of XLH demonstrate beneficial effects of calcitriol supplementation on growth plate maturation and skeletal microarchitecture in absence of phosphate supplementation [49]. In patients with XLH, active vitamin D, in combination with phosphate supplementation, leads to improvements in rickets, limb deformity, growth and gains in height [50,51].
The timing of when PBM is attained has been reported to help define the lifecycle phase for optimal bone health, allowing for appropriate and timely targeting of interventions aimed at achieving optimal PBM [52]. A large age range for the attainment of PBM, BMD and peak of total bone mineral content has been reported in the literature [34-36, 52-54], but in general is reported to be achieved in early adulthood.
XLH is a rare, hereditary, progressive and lifelong renal phosphate wasting disorder caused by loss-of-function mutations in PHEX (phosphate-regulating endopeptidase homolog X-linked). For full reviews refer to Beck-Nielsen 2019 [55] and Haffner 2019 [50]. This condition is primarily inherited in an X-linked dominant pattern, whereas approximately 20-30% of cases arise from spontaneous mutations [55-57].
Although the pathogenesis of XLH is not fully understood, studies indicate that loss of PHEX function leads to the enhanced production, primarily from the osteocytes in the skeleton, of the phosphaturic hormone fibroblast growth factor 23 (FGF23). Elevated levels of serum FGF23 increase renal phosphate wasting by reducing phosphate reabsorption in the proximal renal tubule and increasing urinary phosphate excretion. FGF23 also downregulates one alpha hydroxylase, which curtails the formation of 1,25(OH)2D [58]. The resultant decrease in active vitamin D production impairs phosphate absorption from the gut. These actions, in turn, reduce serum phosphate concentration [59].
Growth retardation is a major manifestation of XLH in children. The abnormal, disproportionate growth is primarily seen in reduced longitudinal growth of endochondral long bones [60]. Diminished growth with declining z-scores has been reported in children with XLH as young as 6 months of age and the growth velocity continues to decline progressively during early childhood, remaining behind that of normal population growth curves [61]. In another study, growth retardation in children under 1 year of age was observed to be similar between boys and girls. However, after reaching the age of 5 years, stunting was more pronounced in boys than in girls [62].
The mechanism responsible for growth impairment in XLH remains to be elucidated. It has been suggested that both cartilage and bone alterations are important underlying causes for the impaired growth and long bone deformities observed in XLH [63]. Moreover, studies using murine models of XLH indicated that antagonizing FGF23 activity, by administering a mitogen-activated protein kinase (MAPK) pathway inhibitor, resulted not only in the acceleration of growth and improvement of rickets, but also in a normalization of growth plate structure [64]. Further studies are needed to clarify the relationship of elevated FGF23 and growth retardation in XLH, but experimental studies suggest that blocking FGF23 action accelerates growth velocity without the risk of worsening bone deformities [17].
Treatment of XLH with oral phosphate supplements and active vitamin D analogues is insufficient to normalize longitudinal growth, although improvements in biochemical parameters, in addition to partial prevention or amelioration of rickets and other manifestations of the disease have been reported [50,62,65,66].
The significant morbidity associated with XLH that impacts bones, joints and muscle, in addition to dentition and hearing, is well documented [3]. The resultant, progressive musculoskeletal abnormalities throughout life are debilitating - causing pain and stiffness, impairing mobility and physical function, decreasing range of motion and reducing health-related quality of life [67].
XLH is primarily characterized by chronic hypophosphataemia, which in addition to other disease factors such as changes in tissue non-specific alkaline phosphatase, pyrophosphate, calcitriol, the direct impact of FGF23 and compromised activity of the PHEX protein, exerts deleterious effects on bone development. These include reduced longitudinal growth, impaired bone mineralization, osteoid accumulation leading to osteomalacia, and an increase in skeletal osteopontin deposition contributing to the local inhibition of bone mineralization [3,68]. In XLH, apoptosis of the hypertrophic chondrocytes is halted, followed by a decrease in chondrocyte proliferation and loss of organization of the proliferative columns, resulting in reduced and delayed bone growth at the growth plate [69].
Hypomineralization of newly formed bone leads to accumulation of osteoid and weakened bones that may undergo deformation from applied strain and forces on the body [3]. Deformities in the lower limbs become more apparent when the child starts to ambulate [3]. Of note, mild lower limb extremity bowing has been reported in infants with XLH who may not be fully ambulatory [70]. Deformities of the weight-bearing long bones of the lower limbs include genu varum and genu valgus, with associated gait abnormalities and a typical waddling gait, and reduced muscle function [71]. Lower extremity torsion and rotation of the long bones may also be observed [72].
Upper limb deformities may be observed when the child starts crawling (Beck-Nielsen S, professional experience). Loading appears to affect the function of hypomineralized growth plates, which collectively cause leg length to be more affected than arm span in patients with XLH [3,73].
Bidirectional axial transmission (BDAT) bone ultrasound of the radius and tibia of children and young adults with XLH aged between 4.2 and 20.8 years revealed a poorer bone quality in terms of matrix stiffness and strength compared with healthy controls [74]. Trabecular thickness in the tibia was significantly higher in the patients with XLH compared with the control groups. In addition, cortical thickness in the tibia showed a tendency to be higher in patients with XLH compared with the control groups [74].
Moreover, a study using HR-pQCT found that adults with XLH, in addition to having a higher total bone cross‐sectional area of both the tibia and radius, had a reduced cortical thickness and a lower total vBMD [75]. Surprisingly, given that XLH is a disorder that impairs bone mineralization, patients with XLH were found to have a reduced risk of fractures compared with normal reference data [73]. Reasons for this observation and normal bone strength estimates are speculated to be caused by altered geometry with broader and shorter bones resulting in a positive impact on fracture prevention. Furthermore, the lower risk-taking behaviour may also contribute to the fracture risk reduction in XLH [75].
The importance of the growth phase in bone development to define optimal bone structure and mass is well established [76]. Although the distinct phases of skeletal development in healthy individuals have been studied in detail [7], similar data for patients with pathological growth conditions are scanty. Understanding skeletal development in disorders associated with impaired growth is important to accurately identify growth patterns that deviate from the expected norm. This will assist in determining whether a decrease in growth velocity is a natural cause of a disease associated with growth disturbance, i.e. XLH, at that particular stage of skeletal development, or indicates the need for optimized medical treatment.
At present, there is no universal definition for the end-of-skeletal growth [2]. Broad parameters that have been used in the scientific literature to define growth include growth velocity associated with final height, bone age, epiphyseal plate closure and chronological age.
A growth velocity of < 2 cm/year has been used to estimate final height in a study of girls with Turner syndrome receiving growth hormone therapy [77]. Although a growth velocity of zero signifies epiphyseal plate closure, [78] there is no consensus on the growth velocity value to determine near-final adult height (Table 1).
Table 1: Definitions of final adult height based on growth/height velocity. View Table 1
Growth velocity is considered normal when it is following a percentile line on a standard growth chart. Deviations from normal in growth velocity may occur due to natural causes of early or late puberty. In such cases, the final height is expected to be within the normal range for that child but will be obtained at an earlier or later age compared with normal average age range.
The growth velocity is lower than normal in XLH. A close observation of activity of rickets both by paracrine and radiological evaluation, and the need for treatment adjustments are necessary during the growth phase, especially during phases of rapid growth [50].
Bone age, which represents the degree of secondary ossification in the long and short bones, [92] can be used to derive biological and skeletal maturity [93]. Ossification of the hand has been considered to represent maturation of the entire skeletal system. Bone age reported at end-of-growth varies in the literature [84,94]. End-of-growth definitions varied from beyond 15 years for boys and beyond 13 years for girls [95-97].
The commonest modality used to assess bone age is radiography of the left hand and wrist, which contain many bones, are easy to radiograph and are often part of the non-dominant hand and, therefore, less prone to injury [98]. Other methods to determine bone age include ultrasonography, CT and magnetic resonance imaging (MRI) [92,93]. A recent technological development, known as BoneXpert, uses digital X-ray radiogrammetry to measure cortical BMD of the second to fourth metacarpal joints and has been shown to be an accurate method for determining bone age [99]. Bone-Xpert is suitable for use in the healthy population and is useful to predict bone age and final height in children with short stature [100,101], although its value in XLH is compromised by the altered bone size, and is therefore considered inapplicable.
Longitudinal growth occurs primarily within the long bones at the epiphyseal growth plate as well as in the vertebral bodies of the spine. During childhood, the growth plate matures and the total height of the growth plate decreases, finally closing in late puberty, with complete replacement of bone. The primary determinant for epiphyseal closure is timing of puberty and this step marks the end of longitudinal growth of the long bones and vertebrae and the attainment of final height [15,102]. Epiphyseal plate closure does not happen simultaneously in the skeleton and a study of union at the epiphyses at the knee involving young males/females aged 18-18.9 years showed that complete epiphyseal union of the femur, tibia and fibula occurred in males/females in 12.5%/10%, 0%/10%, and 25%/60%, respectively [20,103,104].
Techniques used to identify time of epiphyseal fusion include the Tanner-Whitehouse 2nd edition (TW2) radius-ulna-short bone, which uses 20 regions of the bone to assess bone age [105]. Each of these regions is further split into several developmental stages, one of which identifies when epiphyseal fusion is complete [106].
During adolescence, the growth occurring in the spine results in an increase in SH, which is not necessarily synchronized with the leg growth [19]. This leads to an increase in SH/height ratio in adolescence indicating that the spinal growth velocity is exceeding the growth velocity of the legs [107]. Although the dimensions of growth in the neural canal, as well as the inter-pedicular distance is nearly complete in early childhood, spinal growth has been reported to continue after skeletal maturity and the cessation of limb growth [19].
Chronological age has been used in the literature inconsistently to define end-of-growth irrespective of the nomenclature used, e.g. adult height, mature height, young adults or final height. The age of 18 years is frequently accepted as the time when the estimated final height is reached [108]. However, the exact age used for determining final stature varies from 18 to 30 years of age, with variations also between gender [95,109,110].
Chronological age to determine final height is seldomly used in isolation to determine skeletal maturity, but rather in combination with other parameters such as bone age and growth velocity to determine end-of-growth more accurately. The Roche-Wainer-Thissen method, as an example, predicts adult stature based on the age, weight, stature, skeletal age and average parental height of a person [111].
The growth course for children and adolescents with slow height velocity and impaired growth, as seen in XLH, is not well characterized in the literature. Despite having a normal birth length, individuals with XLH show impaired growth especially during infancy and early childhood [60,61]. Compared with the Centers for Disease Control and Prevention growth curves, linear height in children with XLH fell behind that of the normal population as early as 6 months of age, and progressively declined during early childhood, with all median height percentiles < 8% between the ages of 2 and 12 years [61]. Results from a multicentre study also revealed that children with XLH from the age of 2 years had a stature mean value that was persistently below -2 standard deviation score (SDS) of normal reference values in healthy children [60].
Applying standard growth charts used for the general population to patients with XLH could likely lead to the underestimation of improvements in children and adolescents with XLH and pathological growth. In XLH management, inaccuracies in measuring or monitoring growth may result in treatment cessation before the full benefits of restoring phosphate homeostasis and achieving final height are realized. Growth curves that are specifically developed for patients with XLH will shed light regarding the onset of reduced growth velocity and help characterize the course of growth from birth to adulthood. A widely accepted definition of end-of-growth, aided by XLH-specific growth charts, will guide optimal management strategies and treatment guidelines.
Disease-specific growth charts are important tools that help to understand the growth pattern and pathogenesis of hereditary diseases associated with growth failure [112]. A precedent for growth curves in diseases affecting growth has already been set. Disease-specific growth charts are available for achondroplasia, sickle cell anaemia, Turner syndrome, Williams syndrome and Noonan syndrome [112-114]. Specific growth charts have also been developed for disorders that are associated with height increase in a population, which is the case for patients with Marfan syndrome who often have a tall stature [115]. In addition, growth charts for children with XLH are now available [61]. For many rare syndromes, sufficient longitudinal growth data for varying individual patterns is usually impossible to collate to establish meaningful growth charts. Therefore, current practice also utilizes normal population growth charts with extended SD-lines down to -5SDS to track children with extreme short stature.
As longitudinal growth in the endochondral long bones is largely affected in XLH, and in addition, eventual bowing of the long bones may affect the height measurements, other methods of measuring skeletal growth in pathologic growth conditions may be considered.
When the measurement or interpretation of standing height represents a challenge, especially in the presence of gradually bowing of the lower extremities, arm span length (ASL) can be considered. ASL is a commonly used body parameter for predicting height, [116-118] and has proven useful to identify individuals with disproportionate growth abnormalities and skeletal dysplasia as well as to predict age-related loss in stature [117]. The arm span to height ratio (ASHR) in normal individuals is usually around one, but it differs between children and adults (age-dependent), gender and ethnic groups [118]. ASL may also be useful in monitoring the longitudinal bone growth at a site not affected by weight bearing [117].
The intercondylar (IC) and intermalleolar (IM) distance are measured at the knee or the ankle respectively, using a measuring tape with the patient in a standing position. This measurement is used to assess valgus or varus leg deformities. Performing IC and IM measures at clinical visits will reveal a worsening in a leg deformity.
SH is measured from the vertex of the head to the seated buttocks by a normal stadiometer, with the patient sitting on a stool subtracting the height of the stool from the measured height. The SH can be used instead of stature, if height cannot be accurately measured because of lower limb deformities, revealed by the IC/IM values [107]. A multicentre study involving 76 children with XLH reported that SH/height ratio SDS increased significantly during late childhood/adolescence. The reduction in height SDS in XLH was thus primarily driven by the reduced growth of the lower limbs and is suggestive of a dissociated growth retardation primarily affecting the long weight-bearing bones. The degree of leg bowing was only weakly associated with leg length [60].
The absence of a universally accepted single definition for a growing skeleton poses a challenge for the assessment of the end-of-growth in young adults.
A definition that may be adapted to be applicable to disorders affecting bone growth and bone shape will raise awareness on disease-specific growth patterns and address the possible pitfalls when interpreting height data. It will also aid in the assessment of new therapies and the monitoring of the response to new and existing therapies. The understanding and definition of a growing and maturing skeleton may be divided into two modalities:
Individuals affected by disorders that impair skeletal growth and development exhibit a growth pattern that deviates from and falls behind that of standard growth curves. The growth pattern may even be dissociated with different body segments variably affected, such as in XLH. Thus, there is a need for growth curves specifically for XLH to identify growth patterns that deviate from the expected normal growth in XLH.
Underestimation of growth improvement in patients suffering from pathological growth, i.e. when longitudinal growth goes unrecognized due to progressive deformation of the lower extremities, could lead to treatment cessation before the full benefits of therapy are realized. The use of alternative measures such as SH or ASL should be considered when estimating growth or the cessation of growth in diseases characterized by deformations of the spine or lower extremities. In XLH, the use of ASL or SH is independent of eventual bowing of the lower extremities and the lower growth velocity of the weight-bearing long bones.
Thus, in the clinical setting a proposed definition of final height achievement:
1. The increase in height of < 0.5 cm/6 months in two consecutive measurements.
2. The radiological closure of the growth zones at the wrist and knee supports the decision for obtained final height.
1. The increase in height of < 0.5 cm/6 months in two consecutive measurements, when there is no worsening of eventual deformation of the lower extremities and no worsening of an eventual scoliosis.
2. If there is worsening of deformation of the lower extremities and/or worsening of an eventual scoliosis, utilize the increase in arm span of < 0.5 cm/6 months in two consecutive measurements.
3. The radiological closure of the growth zones at the wrist and knee supports the decision for obtained final height.
Continued bone modelling after final height has been achieved:
Providing a clear measure of when bone modelling/appositional growth has reached its maximum is more complicated, given that it continues into adult life albeit at a slower rate. In healthy individuals, skeletal maturation can be estimated by the maximum value of bone area revealed by DXA, where normative data for comparison is available. In diseases affecting bone area, the use of DXA interpreting bone area, where the patient serves as their own control may be a suggestion, but due to the size artefact caused by wider bones in XLH, a comparison with normal data is not applicable [119,120].
Achieving PBM:
PBM, as the maximal value of bone mass is usually expressed as the maximum value of total body BMC (TBMC) or total body BMD (TBMD) for a normal individual [36]. The direct measurement of when PBM is reached is challenging in diseases affecting bone area. DXA measures of both TBMC/TBMD is again confounded by diseases affecting bone area, where the wider bones in XLH makes comparison with normal DXA data inaccurate. Estimating vBMD by HR-pQCT or CT will eradicate the confounding size artifact, providing a true bone density measure in diseases affecting bone area.
A proposed definition of a maturing skeleton is:
A skeleton is still considered to be maturing when it is accruing bone mass and bone density.
Thus, the skeleton continues maturation after longitudinal growth has ceased.
There are many modifiable and non-modifiable factors that can influence the growth and maturation of the human skeleton. Disturbances in physiological processes that impair normal skeletal development during childhood can lead to lifelong consequences such as retarded growth and numerous bone-related deficits. XLH is an example of a hereditary, rare metabolic bone disease that is characterized by chronic hypophosphataemia. Phosphate insufficiency during the pivotal period of bone development in early life leads to rickets and poor longitudinal growth, resulting in limb deformities and short disproportionate stature.
The lack of a universal definition of a 'growing skeleton' poses a challenge in assessing end-of-growth in young adults in general. Several working sessions were held with the authors to develop a standard definition of a growing and maturing skeleton that would be applicable to both healthy and pathological skeletal growth.
The authors propose, in the clinical setting, a definition of final height achievement in children with a normal growth pattern. An adapted definition is also proposed for XLH and other diseases that affect skeletal growth, bone shape and development. A definition for a maturing skeleton is proposed as being one that is still accruing bone mass and bone density; this implies that skeletal maturation occurs after cessation of longitudinal growth.
These definitions may be beneficial to guide assessment of end-of-growth in young adults, in both health and skeletal disease. Moreover, the proposal of a definition of final height achievement addresses the pitfalls when interpreting height data in young adults with disorders affecting bone growth and bone shape.
Dr. Beck-Nielsen reports research grants from Kyowa Kirin, receiving payment for invited speeches for Kyowa Kirin, Novo Nordisk, and PharmaCosmos, and consultancy provided for Kyowa Kirin. Dr. Greggio reports receiving payment for invited speeches for Kyowa Kirin, and consultancy provided for Kyowa Kirin. Dr. Hagenäs reports receiving payment for invited speeches for Kyowa Kirin, and consultancy provided for Kyowa Kirin. All authors state no relation for their disclosures to the submitted work.
This manuscript was based on a series of meetings organized and sponsored by Kyowa Kirin International. Medical writing support was provided by Sciterion and funded by Kyowa Kirin International.
These authors contributed equally to this work.