Background: Fibrodysplasia Ossificans Progressiva (FOP) is an extremely rare and disabling disorder that affects 1 in 2 million individuals worldwide. It is caused by mutations in bone morphogenetic protein which leads to extra-skeletal ossification of soft tissues in a characteristic cranio-caudal pattern. Hallux valgus, episodic flares and progressive functional disability are characteristic features. The cause of death is often cardio-respiratory failure following thoracic insufficiency.
Methods: This was a retrospective case review.
Results: A two year 5-month-old boy who was otherwise normal from birth presented with a history of painful firm masses over the axilla, back, neck, elbows and occiput associated with hallux valgus and progressive inability to move both upper limbs and the neck. Radiographic features revealed pathognomonic ossification of surrounding connective tissue and histology confirmed muscle replacement by hyaline cartilage and bone. An impression of FOP was made and taper therapy with oral prednisone initiated for management of flares.
Conclusions: FOP is a rare and often underdiagnosed condition. Due to the rarity of this condition, the medical fraternity's awareness of FOP is limited and hence low diagnosis rates might reflect this lack of knowledge. This case is meant to raise awareness of this rare disease in Kenya.
Fibrodysplasia ossificans progressiva, Kenya
Fibrodysplasia ossificans progressiva (FOP) is a rare and disabling disorder with a worldwide prevalence of approximately one in two million individuals [1]. There is no ethnic, racial, gender, or geographic predisposition [2]. It is caused by a heterozygous activation or gain of function mutation in ACTIVIN A receptor or type 1 ACTIVIN LIKE KINASE 2 bone morphogenetic protein [3]. It is characterized by extra-skeletal ossification of soft tissues with episodic flare ups and progressive functional disability [2]. Thus FOP is popularly known as stone man disease [2]. Other features include hallux valgus, a shortened big toe and calcaneal abnormalities [4,5]. The diagnosis is often missed at birth and only recognized in the first decade of life due to progressive ectopic ossification [5].
Disease progression may occur spontaneously or in association with trauma, vaccination, intramuscular injections or biopsy attempts [5]. It is postulated that by the second decade, many individuals begin to lose mobility and by the third and fourth decade most patients are wheelchair bound [5]. Death eventually occurs in the fifth decade or earlier due to cardiorespiratory failure preceded by thoracic insufficiency syndrome and pneumonia [6].
We describe a case of a two year five-month-old boy that was referred to our rheumatology service in July 2019. He presented with painful firm masses of bony consistency over the axilla, neck, occiput and the forehead for seven months. Following the appearance of these masses, he developed progressive inability to lift the arms, flex his neck and had a characteristic stiff posture while walking. The masses had a waxing and waning nature lasting about 4-5 days but with no complete resolution. He was previously treated with antibiotics for lymphadenitis which was diagnosed by biopsy of the neck mass at a different health facility. There was no history of fever or contact with a patient known to have tuberculosis. He was born of a non-consanguineous union and no family history of similar illnesses. He had received all relevant vaccinations but was noted to have delayed speech.
Upon physical examination, the child was noted to be sick looking and irritable. He had bony masses on the posterior aspect of the neck which were tender on palpation and attached to the underlying subcutaneous tissue. He also had similar masses on his anterior and posterior chest wall measuring 1 cm by 1 cm anteriorly and larger masses- measuring 5 cm by 5 cm posteriorly. He had bilateral hallux valgus and both elbows held in fixed flexion. His fingers had varying degrees of fixed flexion. The lower limbs were unaffected.
Laboratory evaluation included a complete blood count, urea, electrolytes, creatinine, and liver function tests, anti-cyclic citrullinated peptide (anti-CCP), rheumatoid factor, C-reactive protein, lactate dehydrogenase, creatinine kinase, serum calcium, phosphate all of which were within normal ranges.
Bilateral forearm radiographs revealed extensive dystrophic soft tissue calcification around both elbows outlining the joint capsules. A chest radiograph revealed similar appearing dystrophic soft tissue calcification in the bilateral axillae. Radiographs of the spine demonstrated extensive "sheet like" soft tissue dystrophic calcifications overlying the posterior elements of the vertebral column separate from the vertebrae (Figure 1).
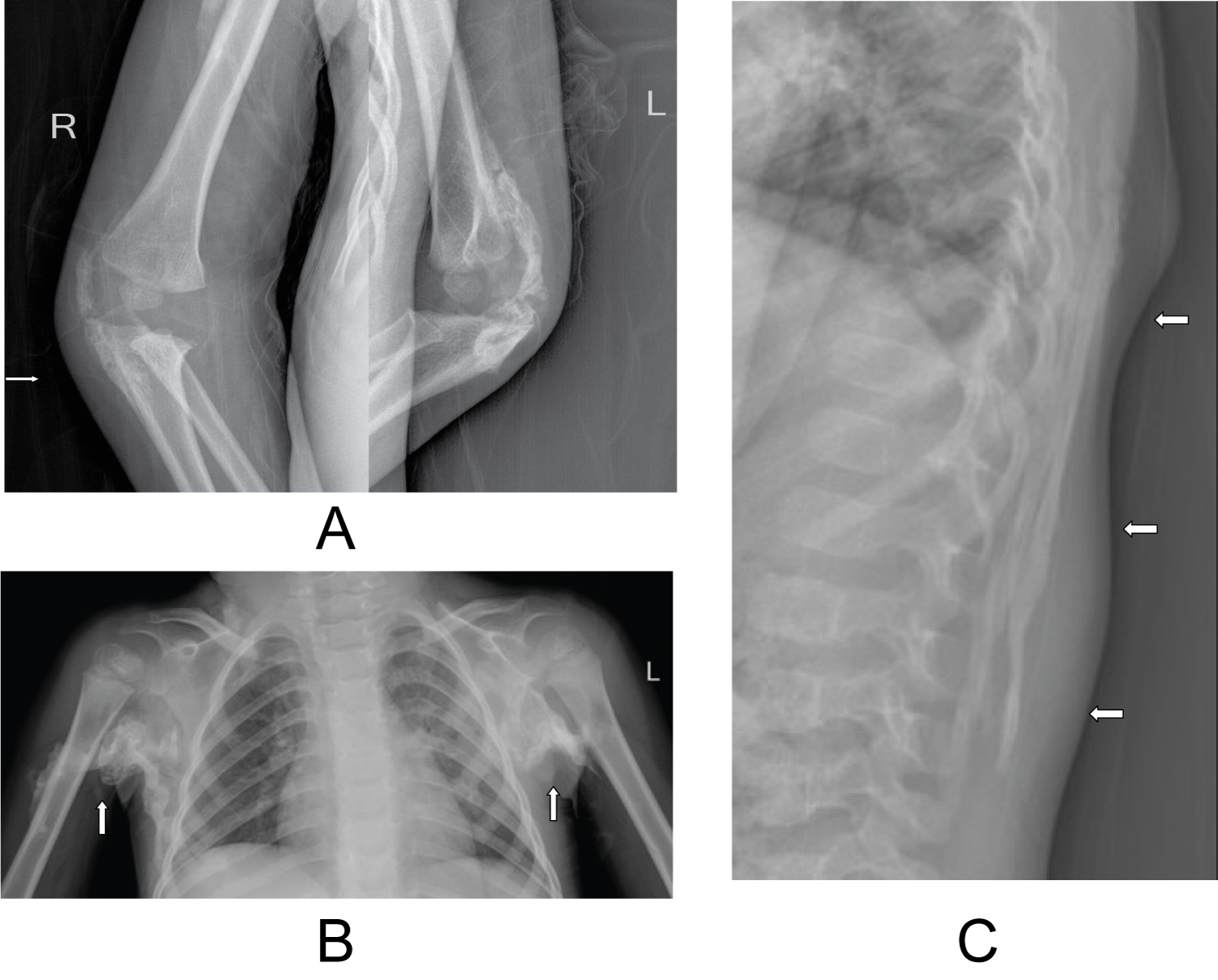 Figure 1: Radiographs illustrating areas of established heterotopic ossification in both elbows, axillae and within the posterior paraspinal soft tissues (white arrows) (a) Radiographs of the right and left elbows; (b) Chest radiograph; (c) Thoracic spine radiograph. View Figure 1
Figure 1: Radiographs illustrating areas of established heterotopic ossification in both elbows, axillae and within the posterior paraspinal soft tissues (white arrows) (a) Radiographs of the right and left elbows; (b) Chest radiograph; (c) Thoracic spine radiograph. View Figure 1
An ultrasound guided core biopsy of the posterior chest wall mass was performed under sedation using a 16G Bard Magnum biopsy needle (Bard Medical, Georgia, USA). Histology showed muscle replacement by hyaline cartilage and bone, with intervening fibromyxoid stroma (Figure 2).
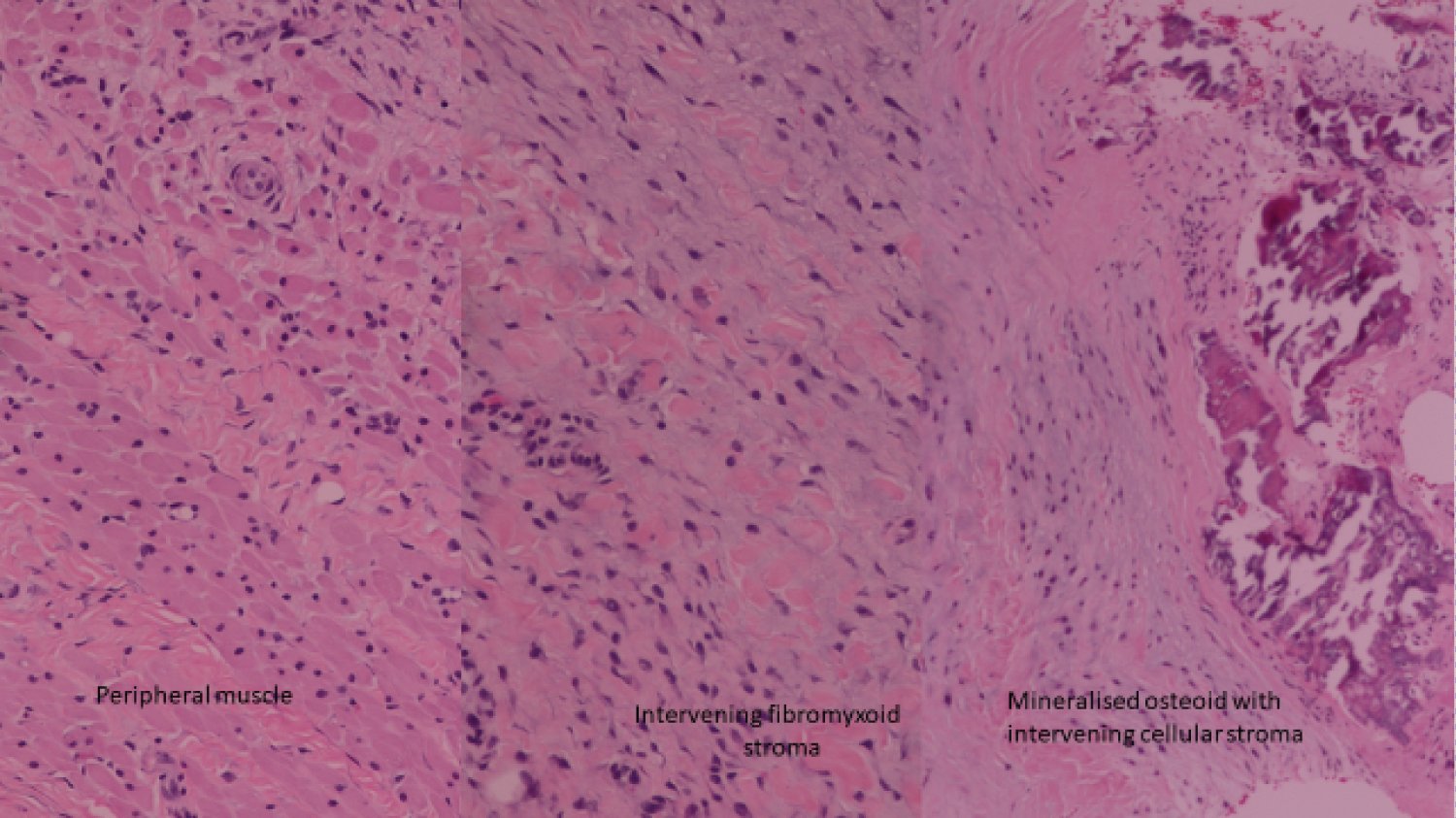 Figure 2: Histology showed muscle replacement by hyaline cartilage and bone, with intervening fibromyxoid stroma. View Figure 2
Figure 2: Histology showed muscle replacement by hyaline cartilage and bone, with intervening fibromyxoid stroma. View Figure 2
Based on this finding, clinical presentation, histological findings and characteristic radiographic features a diagnosis of fibrodysplasia ossificans progressiva was made and the child was discharged home on non-steroidal anti-inflammatory medication for pain and a taper dose of steroid to manage the ongoing flare. He is currently undergoing physiotherapy and occupational therapy and parents have been advised on the triggers for flare ups. The family was enrolled into the international FOP support group and scheduled to have a follow up hearing and speech assessment.
We present a 2 year 5-month-old boy that had heterotrophic ossification in the axilla, neck, back, chest and anterior elbows for 7 months prior to receiving a confirmatory diagnosis of FOP despite several outpatient visits highlighting the importance of raising awareness of FOP among the medical fraternity in Kenya. Fibrodysplasia ossificans progressiva (FOP) is a progressive heterotopic endochondral ossification (HEO) disorder first described by Guy Putin in 1648 [7].
Children are generally normal at birth except for subtle signs like hallux valgus which is a clinical hallmark for the disease as was the case in our patient [5]. Other features include short, malformed thumbs, clinodactyly, neck stiffness, hearing loss and sporadic painful episodes of rapidly progressive bony masses which is pathognomonic of "flare-ups" [6,7]. Progression tends to be cranial-caudal, dorsal-ventral, axial-appendicular, and proximal to distal with sparing of the diaphragm, tongue, extra-ocular muscles, cardiac muscles, and smooth muscles [8,9]. The heterotrophic bone eventually extends across joints, resulting in progressive and irreversible immobility, thoracic insufficiency and death due to cardiorespiratory depression or pneumonia [6]. Minor trauma such as intramuscular immunizations, falls or influenza-like viral illnesses can trigger painful new flare-ups [8]. In our patient, we suspect the frequent falls while playing was the trigger for his flare-ups.
Routine biochemical evaluations are usually normal, although the serum alkaline phosphatase activity and the erythrocyte sedimentation rate (ESR) may be increased, especially during disease flare-ups [10]. C-reactive protein elevation is a more specific test than ESR for monitoring flares but lacks robust data for routine use [10]. Urinary basic fibroblast growth factor levels may be elevated during flares and coincides with the early fibro-proliferative lesions [10]. On the contrary, our patient had a normal CRP and alkaline phosphatase activity.
Radiological investigations reveal normal modeling and remodeling of the heterotopic skeleton [8]. Histologically, early FOP lesions contain an intense mononuclear and perivascular infiltration of macrophages, mast cells, and lymphocytes [11-14]. Following a rapid and destructive inflammatory stage, there is an intense fibro-proliferative phase associated with robust angiogenesis and neovascularity [11-14]. Similarly, biopsies taken from our patient revealed mineralized osteoid with intervening cellular stroma.
The misdiagnosis of FOP approaches 90 per cent of affected individuals worldwide [10]. Children often undergo unnecessary and harmful diagnostic biopsies that exacerbate the progression of the condition [10]. However, in our patient, given that there were concerns of a bone malignancy, a biopsy of the mass was performed. The high rate of misdiagnosis of FOP may be due, at least in part, to the inadequate descriptions and publications of FOP [10].
Definitive genetic testing of FOP is now available and can confirm a diagnosis of FOP prior to the appearance of heterotopic ossification [10]. Clinical suspicion of FOP early in life on the basis of malformed great toes can lead to early clinical diagnosis, confirmatory diagnostic genetic testing (if appropriate), and the avoidance of harmful diagnostic and treatment procedures [10].
Across Africa, multiple cases of FOP have been reported. In northern Tanzania, the case described, unlike our patient, presented with a unilateral hallux valgus. ACVR1c.617G > A mutation was confirmed in this patient [15]. Due to financial constraints, genetic testing was not undertaken in our case. Multiple cases have been reported from South Africa and one from Mali but none of these authors present an effective treatment regimen that was successful in their patients [13-15].
Hearing impairment occurs in about half of FOP patients [10]. The onset is usually in childhood or adolescence with slow progression during the lifetime [10]. Hearing loss is often conductive, possibly due to middle ear ossification; in some patients, the hearing impairment is neurologic [10]. Hearing aids are often helpful [10]. Learning disabilities and special needs should be addressed by appropriate specialists [10].
Patients with FOP develop thoracic insufficiency syndrome (TIS) [7]. This life-threatening complication of cardiopulmonary function can cause pneumonia and right-sided heart failure [7]. It is associated with costovertebral malformations and orthotopic ankylosis of the costovertebral joints, ossification of intercostal muscles, paravertebral muscles and aponeuroses, and progressive spinal deformity including kyphoscoliosis or thoracic lordosis [7]. Prophylactic measures to maximize pulmonary function and minimize respiratory compromise help to reduce morbidity and mortality from TIS in patients with FOP [7].
While effective treatment for FOP will likely be based on interventions that modulate overactive ACVR1/ALK2 signaling, current management is focused on early diagnosis, avoidance of trauma and optimization of function [10]. Medical intervention is currently supportive [10]. Glucocorticoids are used in managing new flare-ups affecting the function of major joints in the appendicular skeleton [10,16]. Anecdotally, non-steroidal anti-inflammatory medications, leukotriene inhibitors and mast cell stabilizers are useful in management of flare-ups but does not alter the natural history of the disease [10,16]. Trial of bone marrow transplantation, immunosuppressive therapy, surgical interventions have been unsuccessful as treatment [10,16]. We should promote prophylaxis against dental caries and avoid intramuscular injections [7,10,16,17]. In addition, prevention of falls is crucial [7,10,16,17]. Prophylaxis against influenza and pneumonia, as well as measures to prevent respiratory infection and cardiopulmonary complications of restrictive chest well disease, are vitally important to help minimize flare-ups [7,10]. The International Clinical Council on FOP (ICC) was established in 2016 and plans for controlled trials are currently ongoing in the hope of finding a suitable therapy for FOP [14]. Upper abdominal surgery interferes with diaphragmatic respiration and should be avoided if possible [18]. Sleep studies to assess sleep apnea and positive pressure assisted breathing devices such as bipap masks without the use of supplemental oxygen maybe helpful [18].
Patients with FOP have approximately a two-fold higher prevalence of kidney stones than the general population [10]. Immobilization coupled with increased bone turnover is a significant risk factor [10]. As in other susceptible populations, a low fiber diet, deficient water intake, excess animal protein intake, and history of urinary tract infections increase the risk of developing kidney stones in FOP [10]. Extracorporeal shock wave lithotripsy, uteroscopic stone removal, percutaneous nephrolithotomy, and laser lithotripsy have all been used as treatment modalities [10].
FOP, though rare, deserves attention from clinicians. Due to the poor prognosis in these children and the high risk of iatrogenic complications, one must always have a high index of suspicion especially when neonates present with clinical features such as hallux valgus at birth. We hope that this report helps to raise awareness of an often neglected and understudied illness.