Malignant rhabdoid tumor of the kidney is a highly aggressive tumor that affects infants and children. Genetically, these tumors have a deletion or mutation of the SMARCBI/INI gene on chromosome 22q11.2.
We describe a case of renal rhabdoid tumor in a 2-years-old child, who presented with fever and treated surgically.
A 2-years-old child previously healthy, presented with one-month history of on-off fever of unknown origin, clinically, child was irritable, found to have mild congestion in the right ear, enlarge left tonsil, and enlarge left cervical lymph nodes & hepatosplenomegaly. First investigations showed C-reactive protein more than 100 mg/l, child received full course of oral Augmentin. Fever settled down for three days, then restarted again with continuous in nature and high grade. Re-investigation showed C-reactive protein of 120 mg/l, lactate dehydrogenase 432.33 U/L, and WBC 13.30 U/L. Other biochemical & hematological investigations were within normal.
An abdominal ultrasound done and showed hepatosplenomegaly for age, right renal hydronephrosis with echogenic content, mass like lesion.
A pan CT showed fungating lobulated heterogeneously enhancing right renal mass measures 7.319.1 × 7.8, shows areas of cystic degeneration, no calcification, involving renal parenchyma and pelvicalyceal system, extends extrarenal and crosses the midline, inseparable from right psoas muscle, abutting and displacing the aorta, compresses the IVC.
Multiple enlarged porta hepatis and para-aortic LNs, largest measures 2 cm. Left suprarenal hypodense mass measures 12 × 15 mm. Average size of the liver showing homogenous density with periportal edema, with no focal lesions or dilated intrahepatic bile duct.
Normal appearance of the spleen, pancreas, left kidney and suprarenal gland.
No obvious GIT abnormality No free fluid or localized collections.
The final impression of Pan CT was confirmed a picture of fungating infiltrative right renal mass with metastatic lymph nodes, suggestive for rhabdoid tumor of the kidney.
An ultrasound guided biopsy done and histopathology reported as renal parenchyma focally infiltrated by a poorly differentiated malignant neoplasm composed of quite large epithelial cells many of which appear degenerative or crushed possibly related to preceding therapy.
Immunohistochemistry showed focal positivity for CD99 of dubious significance along with focal positivity for EMA, while desmin, MyoD1, Pan- Keratin and CD34 are negative. Also staining for INI-1/SMARCB1 is negative, indicating loss of the product of this gene in the Swi/Snf complex, this is indicating Malignant Rhabdoid tumor of the kidney.
Further staging workup showed normal (brain MRI, bone scan, Echo, BMA) Child begin treated as rhabdoid tumor based on CT findings and biopsy, received 3 cycle of chemotherapy. Then child underwent Right kidney Nephroureterectomy Laparotomy, after that patient completed AREN321, Regimen UH-I, child still under follow up of pediatric hematology and oncology, furthermore the initial assessment showed residual disease (Figure 1).
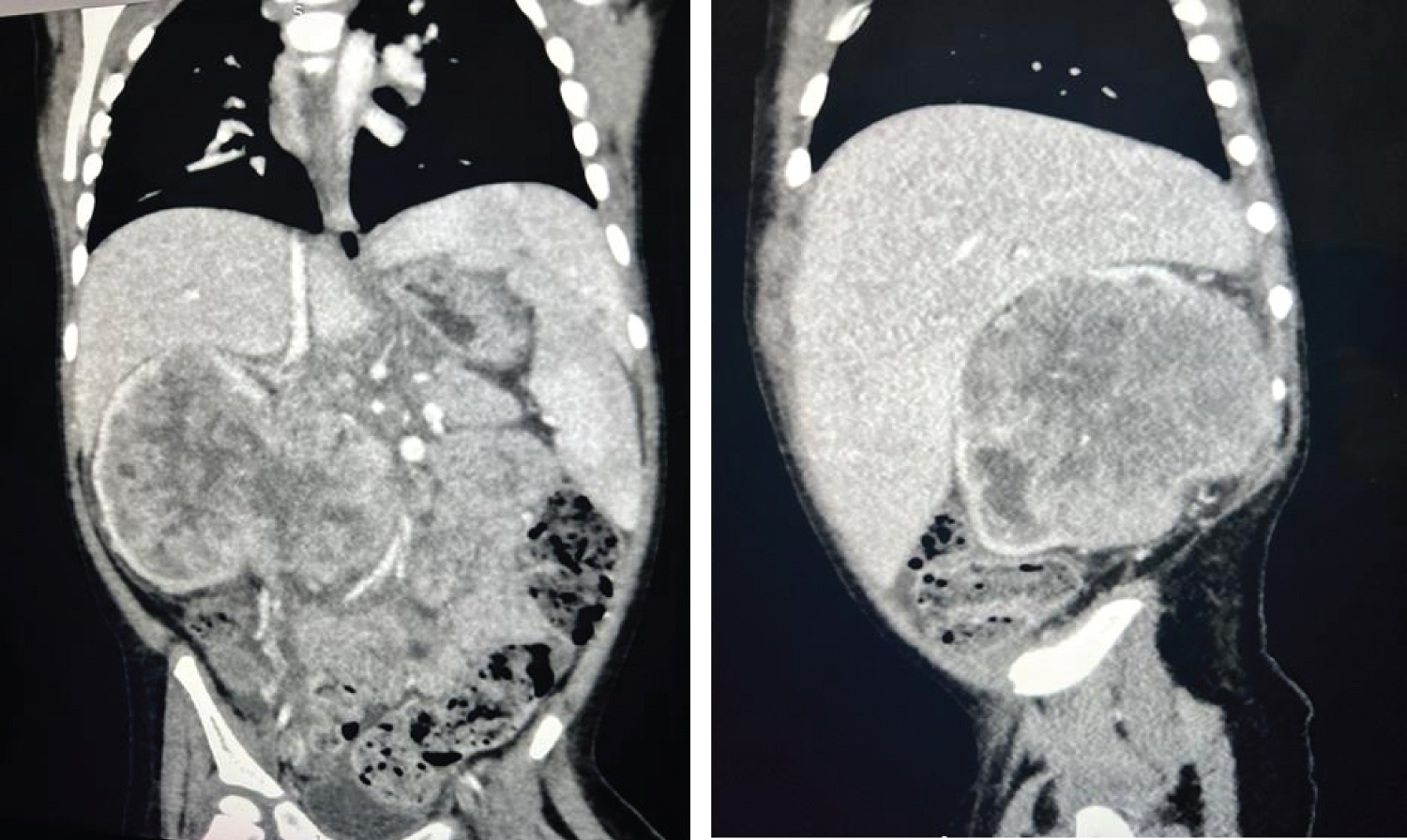 Figure 1: A CT showed fungating lobulated heterogeneously enhancing right renal mass and showed areas of cystic degeneration, involving renal parenchyma and pelvicalyceal system, extends extrarenal and crosses the midline, inseparable from right psoas muscle, abutting and displacing the aorta, compresses the IVC.
View Figure 1
Figure 1: A CT showed fungating lobulated heterogeneously enhancing right renal mass and showed areas of cystic degeneration, involving renal parenchyma and pelvicalyceal system, extends extrarenal and crosses the midline, inseparable from right psoas muscle, abutting and displacing the aorta, compresses the IVC.
View Figure 1
Rhabdoid tumor of the kidney (RTK) is a rare but extremely aggressive pediatric tumor with survival rates ranging between 20% and 25%. It represents 1.5% of all the pediatric cancers of the kidney [1-4].
The particular cell type responsible for Rhabdoid tumor of the kidney is yet unknown. It has been suggested that the origin could be from primitive cells in the renal medulla. Rhabdoid tumors of the kidney consist of cells organized in diffuse sheets, alveolar or trabecular patterns [4,5].
RTK was first identified as a histologic subtype of Wilms tumor in 1978 and was later recognized as a distinct tumor in 1981. Pathologists from the National Wilms' label it as "rhabdoid" because it resembles a rhabdomyosarcoma microscopically, despite the fact that electron microscopy, immunoperoxidase, and cytogenetic investigations revealed no skeletal muscle markers. RTK is a monomorphous tumor, as opposed to Wilms tumor, which is made up of blastemal, stromal, and epithelial cells [1,2,4].
Wilms tumors with good histology account for 75% of new malignancies and high-risk renal tumor as anaplastic Wilms tumor, metastatic renal sarcoma, malignant rhabdoid tumor, and carcinoma make up the remaining 25% [6] .
The International Incidence of pediatric Cancer study's database shows that it accounts for 2% of all pediatric kidney malignancies [4,6].
The tumor arises during the perinatal period, the first year of life, and in certain older individuals. It affects males more than females (1.5:1), with the average age at diagnosis being around 18 months [4,6].
One significant prognostic factor that was previously overlooked is age at diagnosis. Patients diagnosed before one year of age face a poor prognosis and are more likely to develop CNS tumors; those diagnosed after one year of age have a somewhat better prognosis [3,6].
Rhabdoid tumors of the kidney most commonly metastasis to regional lymph nodes and the lungs, although they can also spread to the liver, bone, and brain. Furthermore, there has been a link established between RTK and certain secondary brain cancers [1,2,7].
Common symptoms at presentation include gross hematuria and abdominal masses as well as metastases to the lymph nodes, lungs, peritoneum, liver, and central nervous system. Also, can present with hypercalcemia from elevated serum parathormone levels, abdominal distension or pain, fussiness, and fever [4,6,7].
Gross hematuria was found to be the presenting symptom in 59% of renal rhabdoid patients in a study including 50 individuals. In our patient, hematuria was absent. However, the symptoms that were most concerning were stomach distension, and fever, which our patient had it [6].
There are no imaging features that can distinguish malignant rhabdoid tumor from other pediatric renal tumors. Abdominal ultrasonography may reveal tumoral invasion of the renal vein and/or inferior vena cava in malignant rhabdoid tumors. Doppler ultrasonography or magnetic resonance angiographies are recommended for diagnosis [4].
Malignant rhabdoid tumor is generally seen on abdominal CT as a big, lobulated mass in the kidney's core or periphery. Additional radiological tests may be necessary to detect metastases. Chest CT is recommended due to the high prevalence of metastatic disease from non-CNS malignant rhabdoid tumors in the lungs. A brain CT scan is recommended to rule out synchronous primary or metastatic brain tumors [4].
In histology, this tumor is unencapsulated and has sheets of tumor cells that aggressively overrun the normal nephrons. Vascular invasion is typically substantial. Tumor cells have vesicular chromatin, cherry-red nucleoli, and pink cytoplasmic inclusions. Tumors may mostly consist of primitive indifferentiated small round cells, but closer inspection can reveal isolated foci of cells with diagnostic cytologic characteristics [4].
Lack of immunohistochemical staining for SMARCB1 gene (also known as SNF5, INI-I, or BAF47) support histologic diagnosis of malignant renal rhabdoid tumors. Inactivating both copies of a gene by deletions or mutations at chromosomal location 22q11.23 cause biallelic inactivation of SMARCB1 tumor suppressor gene [4,5,7].
Treatment for renal tumors is multimodal and aligned with the highest risk standards. The optimal treatment is a surgical resection with lymph node dissection, followed by chemotherapy. However, due to the tumor's rarity, the optimal treatment is unknown. Chemotherapeutic medicines such vincristine, actinomycine D, doxorubicin, carboplatin, cyclophosphamide, and etoposide are commonly administered in conjunction with surgery, but no single chemotherapeutic drug or combination is the most effective treatment for this cancer. According to the National Wilms Tumor Study (NWTS), etoposide and cisplatin, etoposide and ifosfamide, or ifosfamide, carboplatin, and etoposide were suggested combinations. The International Society of Pediatric Oncology (SIOP) found that preoperative treatment did not enhance prognosis. Delays in surgical treatment increases mortality [4,6].
Treatment-related complications include protein loss, phosphorus, bicarbonate, electrolyte imbalance, cardiac issues, neurological diseases, and myelosuppression [4].
Despite rigorous therapy, the prognosis is exceedingly poor, with 80-90% death [4].
Renal rhabdoid tumor is an aggressive embryonal tumor with a survival rate of 20-25%. The major treatment option is surgical excision with lymph node dissection. Despite multiple trials using chemicals as adjuvant chemotherapy, there is no uniform protocol for this rare cancer.