Congenital pulmonary airway malformation, Spontaneous resolution
CPAM: Congenital Pulmonary Airway Malformation; US: Ultrasound; TB: Tuberculosis; ANC: Antenatal Care; CT: Computerized Tomography; CCAM: Congenital Cystic Adenomatoid Malformation; BPS: Bronchopulmonary Sequestrations; BC: Bronchogenic Cysts; CLE: Congenital Lobar Emphysema; PPB: Pleuropulmonary Blastoma
Congenital pulmonary airway malformation (CPAM) is an infrequent lung developmental anomaly categorized as part of the broader group of congenital thoracic malformations. Alongside bronchogenic cysts and pulmonary sequestration, CPAM is one of the most prevalent congenital lung anomalies. The malformation arises from abnormalities during embryonic development, leading to atypical bronchial morphogenesis and the development of cystic lesions within the lung tissue [1]. The incidence of CPAM is rare overall but accounts for approximately 95% of congenital cystic lung diseases, with an estimated occurrence of 1 in 10,000 to 1 in 35,000 births [1]. Commonly reported symptoms include infection, hemorrhage, dyspnea, pneumothorax, nutritional difficulties, sudden respiratory compromise and malignant transformation [2]. Surgery remains the cornerstone treatment of symptomatic lesions, but the postnatal management of asymptomatic CPAM remains controversial [3]. Spontaneous resolution of CPAM is an infrequent event, and its mechanisms remain poorly understood, but there are reported cases [4-6]. We report a case of CPAM type 1 in an 8-month-old female child who experienced spontaneous resolution without any surgical intervention.
An 8-month-old female child presented with a one-week history of dry cough, fast breathing, grunting, and fever. She was initially managed for severe community-acquired pneumonia at a primary hospital with IV antibiotics and intranasal oxygen for 2 week, but her respiratory symptoms persisted. A chest X-ray revealed pneumatocele with a possible bronchogenic cyst. The patient was referred to specialized Hospital for further evaluation and management. The patient had episodes of fast breathing and grunting, consistent with respiratory distress. The patient had no known family history of asthma or contact with TB diagnosed patient. She was born from para III mother who didn’t have ANC follow up and delivered at home. She hasn’t been vaccinated and exposed to adequate sunlight. Her developmental history is unremarkable.
On physical examination she awake and alert stable vital signs with intermittent desaturation. Chest examination revealed decreased air entry on left lower lung field with no chest wall deformity. On cardiovascular examination, S1 and S2 were heard, no murmur. Abdominal examination revealed soft abdomen moving with respiration. On Genitourinary system, no costovertebral angle tenderness, well-formed female external genitalia. Musculoskeletal and integumentary examination revealed no deformity no pallor or rash. The nervous examination revealed alert patient with normal motor function.
Initial non-contrast chest CT scan with 3 mm thickness revealed left side multiple cystic lucencies in mid and lower lung zone including retrocardiac location (Figure 1). The cysts were of variable sizes, and the largest cyst was 4.7 cm in diameter and was located at left lower lobe displacing the normal lung parenchyma (Figure 2). There was visible communication with the bronchial system. No soft tissue component was observed. Other mediastinal structures, pleura, and chest wall showed normal findings. There was no evidence of enlarged lymph nodes or mass lesions. The airways and fissures were well demonstrated, and no aggressive osseous lesions were seen. The final diagnosis was consistent with congenital pulmonary airway malformation type 1 (Stocker’s classification).
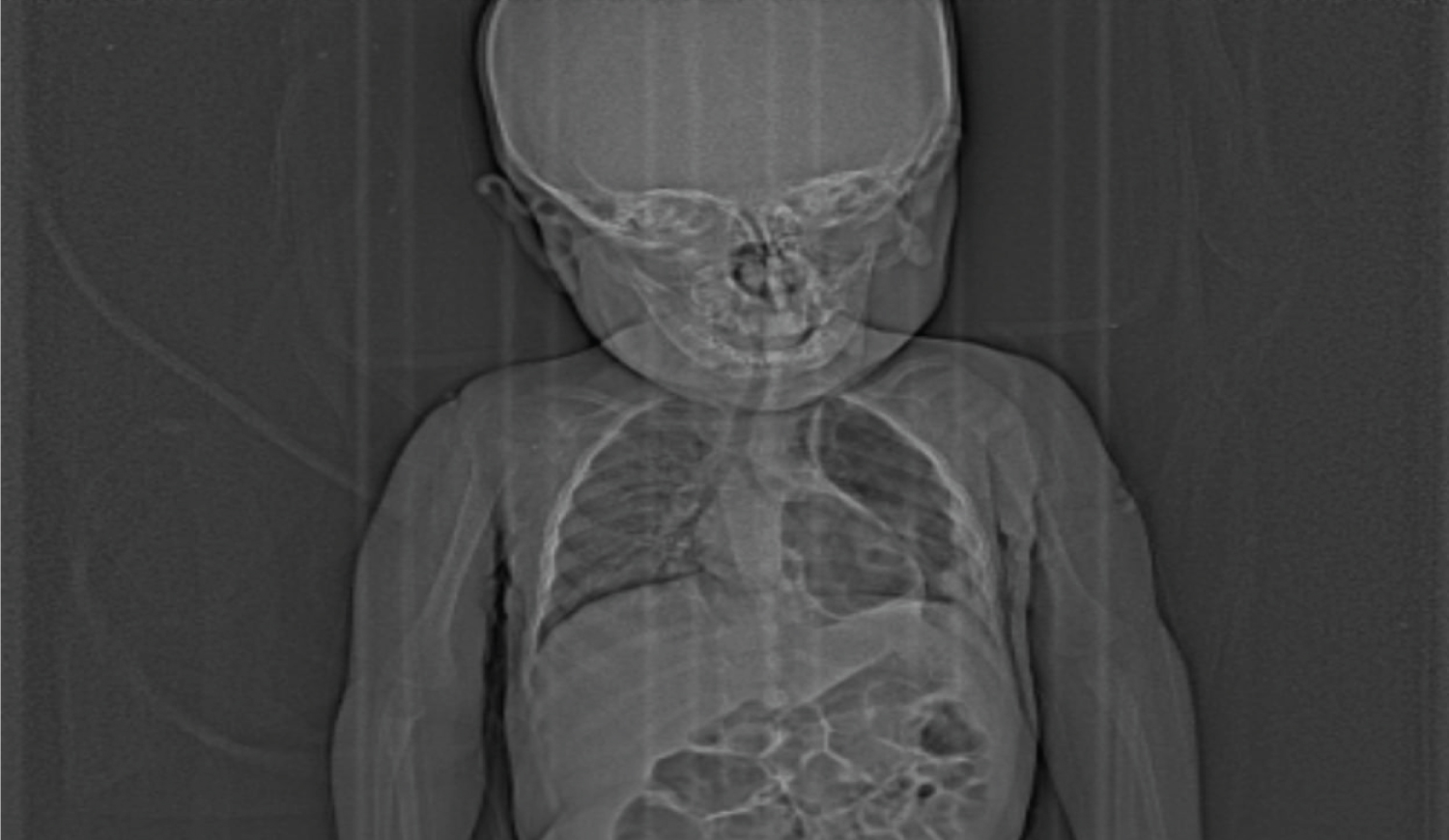 Figure 1: Scout radiography image shows left side multiple cystic lucencies in mid and lower lung zone including retrocardiac location. No significant mediastinal shift seen.
View Figure 1
Figure 1: Scout radiography image shows left side multiple cystic lucencies in mid and lower lung zone including retrocardiac location. No significant mediastinal shift seen.
View Figure 1
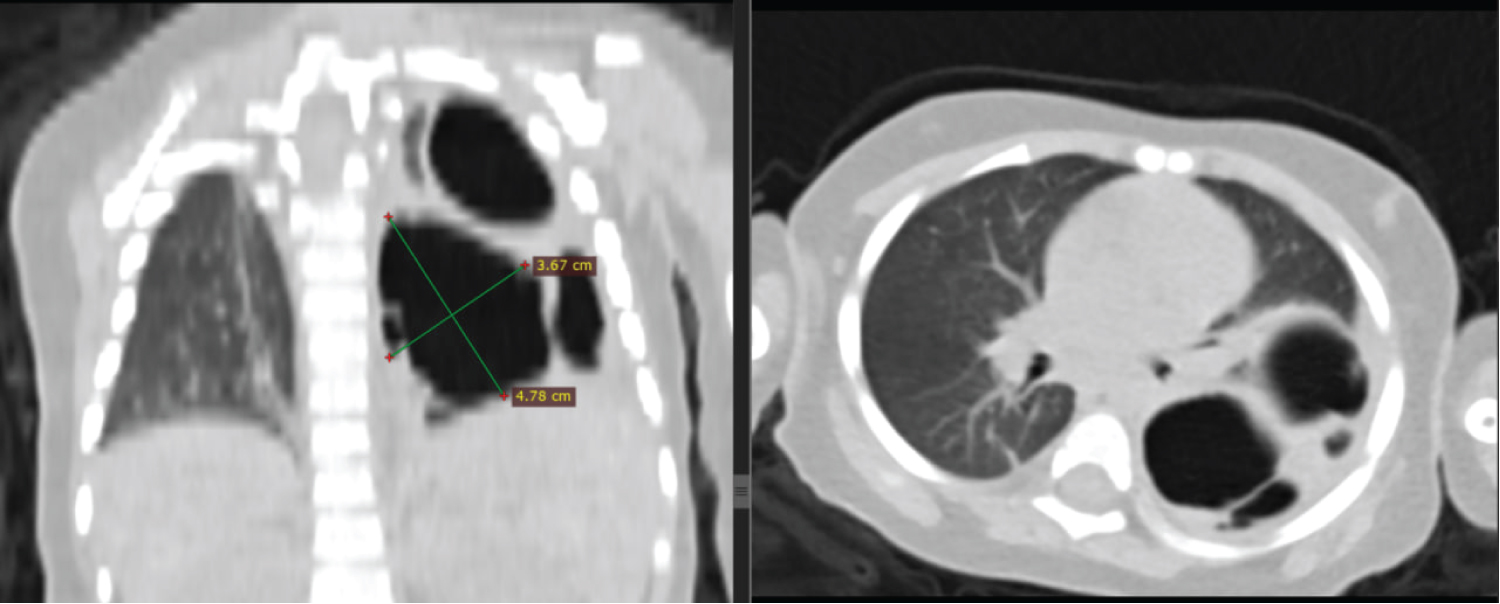 Figure 2: Coronal and axial chest CT scan in lung window shows left lung multiple variably sized cystic lesions, the largest measuring 4.7 cm in long axis dimension. No surrounding consolidation seen. The ipsilateral hemidiaphragm is intact.
View Figure 2
Figure 2: Coronal and axial chest CT scan in lung window shows left lung multiple variably sized cystic lesions, the largest measuring 4.7 cm in long axis dimension. No surrounding consolidation seen. The ipsilateral hemidiaphragm is intact.
View Figure 2
After her presentation patient was managed with supportive care and she started to have clinical improvement within 5 weeks without any specific surgical intervention. The second non-contrast CT scan was taken after 15 weeks and it showed normal lung parenchymal density with no focal or diffuse lesion seen (Figure 3). The pleura were smooth with no effusion or thickening. Mediastinal structures, airways, and chest wall showed normal findings. With this the findings were unremarkable with possible spontaneous resolution of CPAM.
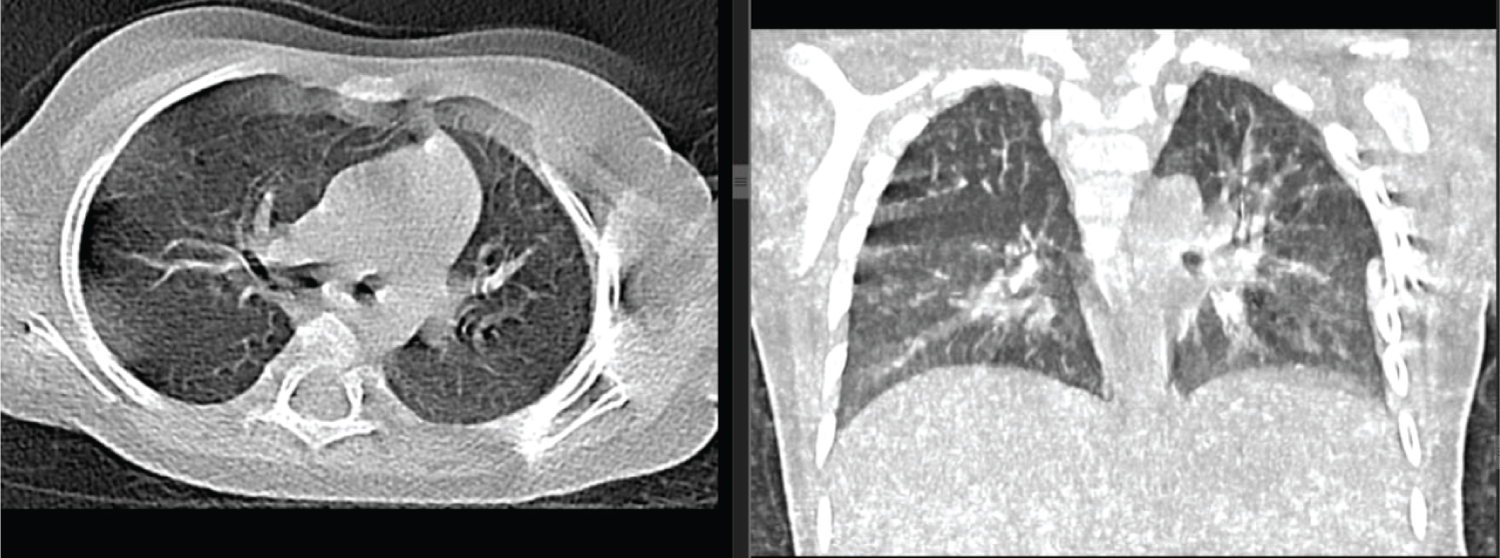 Figure 3: Coronal and axial chest CT scan taken 15 weeks later showed complete resolution of the cystic lesion. No significant volume loss or scarring detected.
View Figure 3
Figure 3: Coronal and axial chest CT scan taken 15 weeks later showed complete resolution of the cystic lesion. No significant volume loss or scarring detected.
View Figure 3
Congenital Pulmonary Airway Malformation (CPAM) is a rare developmental anomaly of the lower respiratory tract, previously known as Congenital Cystic Adenomatoid Malformation (CCAM). Abnormal airway patterning and branching during lung development lead to the formation of lung cysts. The exact cellular mechanisms underlying CPAM are not fully understood, but several genes have been associated with its formation [7].
Two main theories explain the pathophysiology of CPAM: The environmental hypothesis, suggesting genetic defects causing interruptions in lung morphogenesis, and the obstructive hypothesis, which proposes focal air tree obstruction leading to abnormalities. Progress in identifying molecular and gene factors confirms the complex nature of CPAM's pathophysiology [3].
Different classifications have been proposed based on cyst size, cellular composition, and US appearance. Stocker’s classification of CPAM has been modified to include additional lesions and maintain consistency with the original typing of CCAM, which include type 0 and type 4 in the existing type I, II and III [8]. Langston divided CPAM into large-cyst and small-cyst types, while Morotti classified them based on bronchiolar or acinar-alveolar epithelial differentiation. Adzick's classification considers macrocystic and microcystic lesions with prognostic implications. The distinction between histological subgroups is important for predicting outcomes and guiding treatment decisions [7].
Congenital pulmonary airway malformation (CPAM) can exhibit varied antenatal evolution, with cases showing resolution, regression, or progression during gestation. Postnatal diagnosis is often made after pneumonia in patients with no antenatal diagnosis, while wheezing is common in prenatally diagnosed patients not operated on during the first month of life. CPAM is typically detected on postnatal chest X-rays, with contrast-enhanced CT scans being more effective in detecting potential complications [3].
Congenital pulmonary airway malformation (CPAM) has several differential diagnoses, including bronchopulmonary sequestration (BPS), bronchogenic cysts (BC), and congenital lobar emphysema (CLE). Hybrid lesions, which are a combination of CPAM and BPS, have also been identified. Pleuro-pulmonary blastoma (PPB) is a rare, aggressive intrathoracic tumor, often confused with CPAM type 4 due to its rarity [3]. CPAM is now commonly diagnosed prenatally and managed by pediatric surgeons early in life. Surgical management is the consensus for symptomatic patients, and lobectomy is the most prevalent surgical method, although segmentectomy and wedge-resection are becoming safer options.
Spontaneous resolution of CPAM is a rare phenomenon, and the factors contributing to it are not well understood [6]. The rarity of this event underscores the importance of proper diagnosis and monitoring in CPAM cases. Further research is needed to better understand the mechanisms behind spontaneous resolution and its implications for management.
None.
Each named author has equally contributed to the drafting of this manuscript.