Hereditary, isolated Fanconi syndrome has been described a handful of times in the literature, displaying variable Mendelian patterns of inheritance. Some cases segregate as autosomal dominant and may or may not be associated with renal failure, whilst other kindreds show recessive and X-linked recessive inheritance. In 2001, Lichter-Konecki, et al. identified the locus (chromosome 15q15.3) in a kindred with autosomal dominant Fanconi syndrome. In 2014, Klootwijk, et al. found a causal mutation in EHHADH, a gene involved in peroxisomal beta-oxidation resulting in dominant Fanconi syndrome with preserved renal function in a black family.
We applied whole exome sequencing technology to a Caucasian boy presenting with sporadic, idiopathic Fanconi syndrome. We identified a de novo mutation in EHHADH, the same variant as previously described by Klootwijk, et al. This mutation has been observed to cause the protein encoded by EHHADH to mislocalize to the mitochondria, where it interferes with mitochondrial beta-oxidation, the primary source of energy in renal proximal tubular cells. Aberrant mitochondrial beta-oxidation starves the renal tubule cells of energy required to maintain electrochemical gradients, essential for solute reabsorption.
We describe the use of whole exome sequencing to determine the cause of Fanconi syndrome where the family history was compatible with a variety of different modes of inheritance. Our patient is the first Caucasian patient with a de novo mutation in EHHADH causing isolated Fanconi syndrome. This finding allows both informed prognostic discussion and genetic counselling.
Case report, EHHADH, Fanconi syndrome, Whole exome sequencing
Renal Fanconi Syndrome (RFS) refers to a generalised disorder of proximal tubule function resulting in urinary losses of water, electrolytes, low molecular weight proteins, amino acids, phosphate, bicarbonate and glucose. Until recently, the cause(s) of isolated RFS were largely unknown. In 2001, Lichter-Konecki, et al. mapped the locus for a form of autosomal dominant RFS associated with progressive renal failure to chromosome 15q15.3 using genome-wide linkage analysis [1]. The underlying gene has not yet been identified. Magen, et al. described two siblings from a consanguineous family with a recessive form of RFS. The clinical picture was dominated by phosphate wasting resulting from homozygous mutations of SLC34A1, the gene coding for NaPi-IIa, the proximal tubular phosphate transporter [2]. In 2014, Klootwijk, et al. described a family with autosomal dominant Fanconi Syndrome due to a mutation in EHHADH, an enzyme involved in peroxisomal fatty acid oxidation [3]. We describe the presentation and investigation of a child with sporadic RFS with the same mutation in EHHADH.
A two-year-old Caucasian boy born to healthy, unrelated parents was found to have difficulty walking, gross motor delay and short stature. Mild diplegia was suspected but a magnetic resonance imaging scan of his head was normal. At three years of age he represented with genu valgum. Knee radiographs showed evidence of rickets (Figure 1a). Biochemical investigation showed low plasma sodium (131 mmol/L), bicarbonate (13 mmol/L) and inorganic phosphate (0.65 mmol/L) concentrations. Plasma calcium, potassium, lactate and parathyroid hormone concentrations were normal. There was low molecular weight proteinuria (beta-2 microglobulin 108 mg/L (normal < 0.03 mg/L) and retinol binding protein/creatinine ratio 17,909 µg/mmol (normal 3.9-32 µg/mmol)) and generalised aminoaciduria. Tubular reabsorption of phosphate was low at 74%. There was intermittent glycosuria. These results are diagnostic of RFS. The absence of hepatosplenomegaly and cataracts was felt to exclude galactosaemia. The absence of jaundice, hypoglycaemia and a history of vomiting made hereditary fructose intolerance unlikely. The intermittent nature of the glycosuria and absence of hepatomegaly and hypoglycaemia were felt to exclude Fanconi-Bickel syndrome.
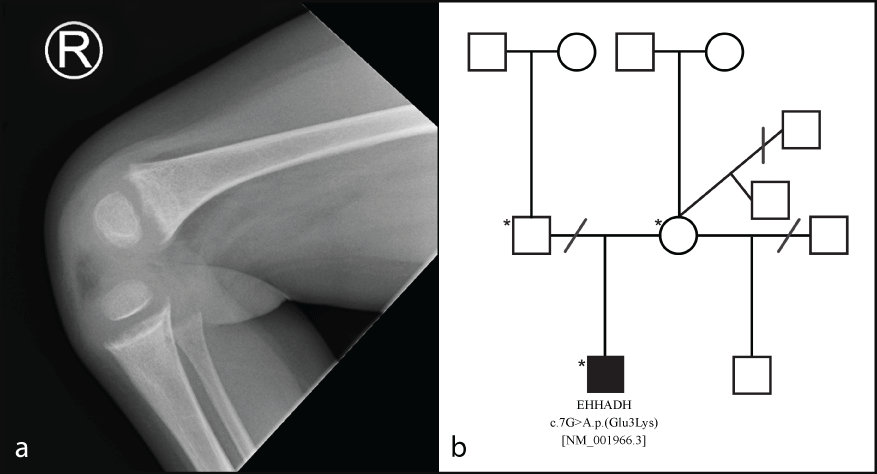 Figure 1: a) A lateral plain film of the right knee of the proband demonstrating clear evidence of rickets. The tibia and femur show metaphyseal fraying and cupping. The distal femur shows slight trabecular pattern coarsening; b) Family pedigree of the proband (affected individual with shaded box). The proband's mother had three male offspring by three different partners. Both half-brothers to the proband are unaffected. Dissolved relationships are denoted by a line through the relationship. An asterisk denotes the three individuals who underwent whole exome sequencing analysis. View Figure 1
Figure 1: a) A lateral plain film of the right knee of the proband demonstrating clear evidence of rickets. The tibia and femur show metaphyseal fraying and cupping. The distal femur shows slight trabecular pattern coarsening; b) Family pedigree of the proband (affected individual with shaded box). The proband's mother had three male offspring by three different partners. Both half-brothers to the proband are unaffected. Dissolved relationships are denoted by a line through the relationship. An asterisk denotes the three individuals who underwent whole exome sequencing analysis. View Figure 1
Further testing was undertaken to identify a cause. White blood cell cystine was normal on two occasions, excluding cystinosis. Plasma caeruloplasmin was 321 mg/L (normal range 210-620 mg/L), excluding Wilson disease. Urinary organic acids showed a large peak of 3-hydroxybutyric acid but no other abnormalities; specifically 4-hydroxyphenyllactic acid and 4-hydroxyphenylpyruvic acids were not elevated, excluding tyrosinaemia. Sanger sequencing of OCRL1 and CLCN5 revealed no pathogenic variants, making Dent's disease and Lowe syndrome unlikely. The mitochondrial m.3243A > G variant was not detected, nor were major mitochondrial DNA rearrangements. A blood spot carnitine profile was normal.
Our patient therefore fell into the category of isolated, idiopathic RFS. The clinical picture did not fit with a defect of NaPi-IIa as he had significant acidosis, a feature absent in the two previously described patients [2]. The family history was largely uninformative showing compatibility with recessive, dominant or X-linked inheritance (Figure 1b).
Whole exome sequencing was undertaken to find a possible genetic cause of RFS. Library capture was performed using Agilent Sure Select V6. DNA extracted from the proband and his two unaffected parents was sequenced at Novogene. Raw data generated from a paired-end sequencing protocol were aligned to the human genome reference 38. A bespoke in-house informatics pipeline was used to call and annotate variants [4]. Variants that did not meet stringent quality control metrics were disregarded from further analysis. Filtering steps exploited parental/offspring segregation and were concentrated in genes and loci previously associated with RFS (GALT, FAH, ALDO8, CTNS, GLUT2, OCRL1, CLCN5, ATP7B, VPS33B, VIPAR, SLC34A1, EHHADH and 15q15.3). Variants were removed from further analysis if synonymous, present with a minor allele frequency > 0.01 from popular database repositories, or with low predicted in silico scores. Post-filtering, one variant remained, a de novo, heterozygous, c.7G > A p.(Glu3Lys) [NM_001966.3] mutation of EHHADH. Sanger sequencing of the parent/offspring trio confirmed the de novo variant. This is the same mutation described by Klootwijk, et al. It results in mistargeting of the peroxisomal enzyme to the mitochondria where it causes mitochondrial dysfunction and RFS [3].
RFS is characterised by a failure to sufficiently reabsorb filtered solutes in the Proximal Convoluted Tubule (PCT), a process that is highly energy dependent and relies primarily on mitochondrial fatty acid oxidation. Indeed, mitochondrial disorders are often complicated by RFS. Adenosine Triphosphate (ATP) is the molecular unit of energy that is essential for maintaining an electrochemical gradient across many plasma membranes; beta-oxidation produces substrates that feed into downstream pathways resulting in the production of ATP. In the PCT, reabsorption of the majority of solutes, as well as water, is dependent on sodium reabsorption which, in turn, is driven by basolateral Na+/K+ ATPase. Therefore, reduced availability of ATP results in urinary losses of water, electrolytes, amino acids, glucose, phosphate, bicarbonate and low molecular weight proteins leading to systemic acidosis, rickets, osteomalacia and short stature.
RFS has numerous aetiologies including inborn errors of metabolism, acquired causes such as drug toxicity and heavy metal poisoning, and mitochondrial diseases. Very rarely, idiopathic inherited forms of RFS have been described. Of the kindreds reported in the literature, disease segregation has been observed to be autosomal dominant, autosomal recessive and X-linked recessive and to occur with and without renal failure [5,6]. In 2001, Lichter-Konecki, et al. genotyped a Wisconsin family with 10 affected individuals presenting with an autosomal dominant form of RFS associated with progressive renal failure. Using linkage and haplotype analysis, the authors mapped the disease locus to chromosome 15q15.3.1
In 2014, Klootwijk, et al. applied genome-wide linkage analysis to a five-generation black kindred with autosomal dominant RFS without renal failure originally published by Tolaymat, et al. [7]. Linkage analysis identified a significant locus on chromosome 3q27 (LOD score > 3). Following the sequencing of all coding genes in the region of interest, a novel heterozygous mutation in EHHADH was found to segregate in all affected individuals [3]. All nine affected individuals in this kindred had normal glomerular function for at least the first six decades.
EHHADH (enoyl-coenzyme A hydratase/L-3-hydroxyacyl-coenzyme A dehydrogenase) encodes a peroxisomal protein, highly expressed in the kidney and liver. Wild-type EHHADH is involved in peroxisomal beta-oxidation of fatty acids. Both mitochondria and peroxisomes have distinct beta-oxidation pathways essential in the metabolism of fatty acids; they utilise organelle-specific enzymes and fatty acid substrates [8]. Peroxisomal beta-oxidation accepts very long chain fatty acids, branched chain fatty acids, bile acid intermediates and long chain dicarboxylic acids as substrates. Contrastingly, mitochondrial beta-oxidation metabolises short chain, medium chain and long chain fatty acids. The end product of beta-oxidation is Acetyl Coenzyme A (acetyl CoA) that fuels the Tricarboxylic Acid (TCA) cycle and ketogenesis. Furthermore, two reduced cofactors (flavin adenine dinucleotide and nicotinamide adenine dinucleotide) are directly used in oxidative phosphorylation to enable ATP production [9,10].
EHHADH shares homology with the α subunit of the Mitochondrial Trifunctional Protein (MTP), a protein dominant in mitochondrial energy production. The mutation of EHHADH c.7G > A p.(Glu3Lys) has been shown to result in a mitochondrial import sequence in the N-terminus of the protein that mistargets it to mitochondria [8]. A stable renal proximal tubular cell line expressing human EHHADH with the c.7G > A mutation showed that mutant EHHADH mislocalized to mitochondria as well as remaining in peroxisomes, whilst the wild-type protein was only detected in peroxisomes. Mistargeted mutant EHHADH is able to replace one or more of the α sub-units of the hetero-octameric MTP thus impairing mitochondrial beta-oxidation of long chain fatty acids [8]. This mutation thus behaves as a dominant negative allele, directly interfering with and impairing mitochondrial function. Transfection studies have shown the mutation to disrupt transepithelial fluid transport in renal tubular cells, in addition to impairing ATP production. This dominant negative model is supported by studies concluding that EHHADH knockout mice are viable and fertile, with apparently preserved mitochondrial function sufficient for adequate growth and development. Moreover, no glycosuria, phosphaturia or aminoaciduria was observed in the knockout mice, nor elevated levels of glycolytic intermediates [3,8].
The direct result of the EHHADH c.7G > A p.(Glu3Lys) mutation is that of defective beta-oxidation in mitochondria that significantly depletes substrates required for oxidative phosphorylation and causes the accumulation of intermediate metabolites e.g. acylcarnitine and hydroxyacyl products. It is of note that our patient had a large peak of urinary 3-hydroxybutyric acid, supporting an aberration in the mitochondrial beta-oxidation pathway, with a skew towards ketosis. Since PCT cells rely so heavily on mitochondrial fatty acid oxidation to maintain the energy-dependent electrochemical gradient required for solute reabsorption, it is easily conceivable how dysregulation of this pathway can lead to RFS. In the only previous report of RFS due to the same mutation of EHHADH, glomerular function was well preserved. There are therefore grounds for optimism regarding the outlook for renal function in this patient [3]. Furthermore, dominant negative mutations are better therapeutic targets than loss of function mutations, offering the potential for personalized specific gene-therapy [11].
To the best of our knowledge, this is the first description of RFS caused by a de novo mutation of EHHADH, and the first reported case in the Caucasian population. Isolated RFS is rare, but in affected patients the diagnosis of this mutation allows for accurate genetic counselling. We might have expected that our patient would harbour a mutation in the 15q15.3 locus, given most reported cases of autosomal dominant RFS have been associated with progressive renal failure [5]. The discovery of the c.7G > A p.(Glu3Lys) mutation in EHHADH therefore has important implications for genetic counselling and disease prognosis. Although the EHHADH mutation is predicted to cause accumulation of acylcarnitine [8], the normal blood spot carnitine profile in our patient suggests that this will not be a useful diagnostic test, possibly because the increase in plasma carnitine is minor and the test lacks the necessary sensitivity.
EGS was funded by the Kerkut Charitable Trust.
The authors declare no conflict of interests.