Systemic mastocytosis is a rare disorder characterized by clonal proliferation, which results in abnormally high numbers of mast cells in the skin, bone marrow, and internal organs, such as liver, spleen, or lymph nodes. Mast cell leukemia and subvariants were stratified by the World Health Organization in 2016, as a subtype of systemic mastocytosis.
Here, we present a case of acute mast cell leukemia that was imatinib-sensitive, with well-known mutations in the oncogene, KIT (c.1565 T > G p.Phe522Cys), and in the splicing factor gene, SF3B1 (c.2098 A > G, p.Lys700Glu). Molecular cytogenetic studies showed that 26% of cells harbored the 46,XX,der(5)t(5;6)(q35;q24),t(10;16)(q26;q23) karyotype abnormality, confirmed with the fluorescence in-situ hybridization technique. Additionally, the patient showed severe hepatosplenomegaly with multiple retroperitoneal, mesenteric, and axillary adenopathies. Although the prognostic survival of patients with mast cell leukemia is around 6 months, this rare case, with a low-frequency (< 5%) p.Phe522Cys KIT mutation, was associated with complete remission and negative minimal residual disease, which lasted for 2.5 years; moreover, the mutation was not detectable after imatinib treatment. When the patient relapsed, the two mutations studied in KIT and SF3B1 genes reappeared, and a new clone emerged with the variant c.2465 A > C p.Asn822Thr, located in the activation loop of the KIT protein.
Molecular analyses of the KIT gene were essential for selecting the appropriate tyrosine-kinase inhibitor. Accurate drug selection can prevent possible resistance and facilitate adequate treatment of subsequent relapses.
Systemic mastocytosis, Mast cell leukemia, KIT, Imatinib, Atypical mast cell
The World Health Organization classification of tumors that arise in hematopoietic and lymphoid tissues represents the worldwide consensus on hematologic malignancies. Mastocytosis is one of the eight subcategories of myeloproliferative neoplasms [1]. Mastocytosis is characterized by a clonal, neoplastic proliferation of mast cells that accumulates within various organs, mainly skin. The major criterion for the diagnosis of systemic mastocytosis is the presence of multifocal clusters of mast cells in bone marrow or other extracutaneous organs [1]. To establish a correct diagnosis, it is necessary to study the immunophenotype and the morphology of mast cells [2]. In addition, molecular analyses are crucial for a definitive determination of the appropriate treatment.
Mastocytosis are rare diseases; the estimated world-wide prevalence is 0.5-1 cases per 20,000, without gender predominance. Systemic mastocytosis (SM) can be benign, like indolent systemic mastocytosis (ISM), or malignant, like mast cell leukemia (MCL). An intermediate form of SM is called smouldering systemic mastocytosis (SSM), which was initially described as a sub-entity of ISM. Other forms include SM with an associated hematologic neoplasm (AHN) and aggressive systemic mastocytosis (ASM) [1]. Patient prognosis varies among the different classes of SM. Indolent forms of mastocytosis are associated with a life expectancy similar to that of the general population, but ASM and MCL have worse prognoses. MCL is a rare entity; its incidence is 1% among patients with mastocytosis. To be considered as MCL entity the percentage of mast cells must be ≥ 20%. MCL is further mainly subdivided into two types. In classical MCL, at least 10% of all white blood cells are mast cells; in aleukemic MCL, less than 10% of all white blood cells are mast cells. Furthermore, there is an atypical presentation of chronic MCL when no organ damage is detected. MCL is associated with cytopenia, hepatomegaly, palpable splenomegaly, malabsorption, skeletal lesions, and organ damage caused by local mast cell infiltration [1].
The proto-oncogene KIT (c-KIT, CD117) encodes a transmembrane type III tyrosine kinase receptor that binds to stem cell factor. KIT is mainly expressed in brain, lung, female reproductive tissues, and skin. Mutations present in several KIT domains promote conformational changes in the receptor that lead to ligand-independent activation. Several variants of the KIT gene have been reported, mainly the p.Asp816Val, which is located in the activation loop (A-loop); this mutation confers resistance to imatinib, the most common treatment. Other known variants are imatinib-sensitive, and they mainly carry mutations in the transmembrane domain of the protein. These variants are associated with a well-differentiated SM phenotype; for example, the p.Phe522Cys [3] and p.Met541Leu [4] variants may predispose to pediatric mastocytosis [5], or in adults, they may lead to an aberrant mast cell immunophenotype [6].
Mutations in the splicing factor 3B1 gene (SF3B1) are frequent in myelodysplastic syndromes (MDS) with ring sideroblasts. These mutations have been associated with good outcomes [7]. SF3B1 mutations are sometimes found in patients with ring sideroblasts (< 10%) and chronic myelomonocytic leukemia. Mutational hot spots in SF3B1 include p.Lys700Glu (~50%), p.His662Gln, and p.Lys666Asn [8,9].
The prognosis of MCL is poor; the median overall survival is less than 6 months [10]. Treatment typically involves tyrosine-kinase inhibitors (e.g., imatinib, when p.Asp816Val KIT is not involved), and/or the protein kinase inhibitor midostaurin, regardless subtype of SM, KIT mutation status, or exposure to an earlier treatment [11]. MCL can also be treated with cladribine, interferon, and hydroxyurea, followed by stem cell hematopoietic transplantation (SCHT), in some aggressive forms of the disease [12-16].
We describe a 33-year-old female with a history of two urticarial episodes 6 months prior, which were readily resolved with antihistamines. The onset of SM evolved over 2 months, with constitutional symptoms of weakness, anorexia nervosa, fatigue, and important weight loss. The symptoms were first associated with a cold and rhinorrhea, productive cough, abdominal pain, diarrhea, spontaneous ecchymosis, and worsening of her general condition. An anamnesis showed poor general condition. Palpation revealed small cervical and inguinal adenopathies and hepatosplenomegaly.
Laboratory findings revealed a white blood cell count of 53,300/mm3, hemoglobin at 7.7 g/dL, and platelets at 31,000/mm3. The patient presented disseminated intravascular coagulation, with a low fibrinogen concentration (123 mg/dL), a prolonged prothrombin time, and high levels of D-dimer. Serum tryptase levels were above 200 µg/L.
The bone marrow infiltration comprised 74% hypogranular mastocytes, some with oval nuclei, located granulation, and a vacuolated cytoplasm. Atypical mast cells with dysmyelopoietic features were found in whole blood. These cells showed positive staining with both toluidine-blue and chloroacetate esterase.
Flow cytometry results showed high-size of blastic population cells infiltration in peripheral blood and bone marrow. The internal complexity of the cells represented 72% of the cellularity with variable expression of the antigens CD117++, CD22+, CD45++, CD30+, CD13+, CD33+, CD4+, and CD43+, which suggested atypical mast cells. The bone marrow karyotype analysis displayed an abnormal 46,XX,der(5)t(5;6)(q35;q24),t(10;16)(q26;q23) in 26% of all metaphases with an unbalanced translocation between chromosomes 5 and 6.
Treatment started with administration of cladribine plus 400 mg daily imatinib. A progressive analytical improvement was observed after treatment, with a diminishing total number of white blood cells. Increases were observed in the hemoglobin concentration and the number of platelets; however, 4 days after treatment initiation, the patient developed severe coagulopathy, decreasing serum fibrinogen levels, and elevated D-Dimer levels. Next, the patient presented an intracranial hemorrhage, mainly at the posterior fossa, which led to an acute triventricular supratentorial hydrocephalus with a transtentorial herniation and encephalic trunk deviation. She required a surgical drain, and she remained in the intensive care unit for several days. After high concentrations of fibrinogen were infused, the serum levels stabilized, and the coagulopathy remitted. At that point, disease evolution was favorable, with an expectation of total resolution of the neurologic complication. Later, the patient received ambulatory treatment with a second cycle of cladribine plus imatinib. The level of atypical mast cells in the bone marrow remained below 0.1% during the complete remission by flow cytometry; the immunophenotype was the same as that observed at diagnosis, the p.Phe522Cys mutation was undetectable, and serum tryptase levels decreased to 3.28 µg/L. After the third cladribine cycle, no atypical mast cells were detected in a bone marrow analysis, and flow cytometry confirmed the absence of atypical mast cells above limit of detection of the assay (< 0.002%). Again, the mutation was undetectable, and the genetic alterations disappeared. Serum tryptase levels were also normalized. During the complete remission of the disease it has been discussed to perform allogeneic hematopoietic stem cell transplantation, but the patient denied this option. After 30 months of 400 mg daily imatinib, the patient exhibited new dermal erythematous lesions, and flow cytometry showed 23% atypical mastocytes in the bone marrow, which indicated a relapse of the disease. At that time, the patient started a second treatment regime, with cladribine (0.14 mg/kg/daily) and 600 mg imatinib. After the first cycle of this treatment, the patient exhibited 12% pathological mastocytes. Next, she was switched to midostaurin (100 mg/12 h) treatment, as a compassionate use treatment; however, the patient did not respond, and she died four months later due to disease complications.
At diagnosis, a p.Phe522Cys mutation in the KIT gene was detected with Sanger sequencing in both peripheral blood and bone marrow samples. Other chromosomal rearrangements were studied, but we found no positive results. We extracted DNA from peripheral blood and bone marrow samples collected at diagnosis to build Next-Generation Sequencing (NGS) libraries. We studied a set of 33 genes with mutations known to be involved in several MDS [17]. We found several variants of the KIT gene, including p.Phe522Cys, (previously observed with Sanger Sequencing), and the mutation, p.Lys700Glu, at 48% allelic frequency, in the SF3B1 gene.
To increase panel coverage of the diagnosis samples, and with the aim of discovering new mutated genes, we performed a comprehensive analysis of the patient's exome and compared two peripheral blood samples, one collected at diagnosis and the other collected 14 months after complete remission. We employed the Ion AmpliSeq™ Exome RDY Kit (Thermo Fisher®) to prepare the NGS libraries and perform sequencing. The sequencing yielded an average main depth of 113X (two samples). After aligning to the hg19 genome sequence, we found an average of 51,075 variants. We then applied the following filters: Minor Allele Frequency (MAF) < 0.1, allele ratio > 0.1, and Variant Effects missense and nonsense. This procedure revealed pathogenic variants in several genes, including ACPL2, EPHA5, SLC19A3, and TENM2. The average variant allele frequency (VAF) was 37.0 ± 2.6%. The SF3B1 mutation was confirmed, and the p.Phe522Cys KIT mutation was again quantified, but with better coverage than that used in the previous analysis. The estimated limits of detection for the exome studies were 3% VAF.
Next, we performed ultra-deep sequencing of the two amplicons that covered the mutated regions in the MDS panel. These results allowed us to follow the allelic frequency of these two mutations for 14 months, starting at disease diagnosis. The frequency of the variant allele that harbored the mutations remained below 3.45% (0.46-3.45%) during the complete remission (Table 1). This last experiment yielded coverages of 120,000X for the KIT amplicon and 70,000X for the SF3B1 amplicon. The ultra-deep sequencing of these two amplicons allows us to detect until 0.1% VAF.
Table 1: New generation sequencing (NGS) results from a molecular biology study of patient specimens. View Table 1
After 30 months of complete remission, the patient relapsed. At that time, we used the NGS MDS panel to analyze four peripheral blood samples, collected two months before relapsing, at the time of the relapse, and one and two months after the relapse, at the time she started the new midostaurin treatment. In addition, we performed the exome analysis that we carried out before with peripheral blood. In this exome analysis, we included two samples: One collected 2 months before the relapse, and the other collected at the time of relapse. These analyses revealed 37,393 variants (average). The allelic frequencies for the SF3B1 and KIT mutations were 11.8% and 7.7%, respectively, at the time of relapse (1000X coverage) and 14.1% and 10.4%, respectively, when we evaluated the exome output NGS data for the same mutations (70X coverage). The exome data analysis confirmed the same pathogenic variants observed at diagnosis (ACPL2, EPHA5, SLC19A3, and TENM2 genes) with a VAF of 13.3 ± 1.5%.
On the other hand, a new p.Asn822Thr mutation (VAF 11%) was observed only at the time of relapse. The variant allele frequency of the mutations and the number of atypical mastocytes are displayed chronologically in Figure 1.
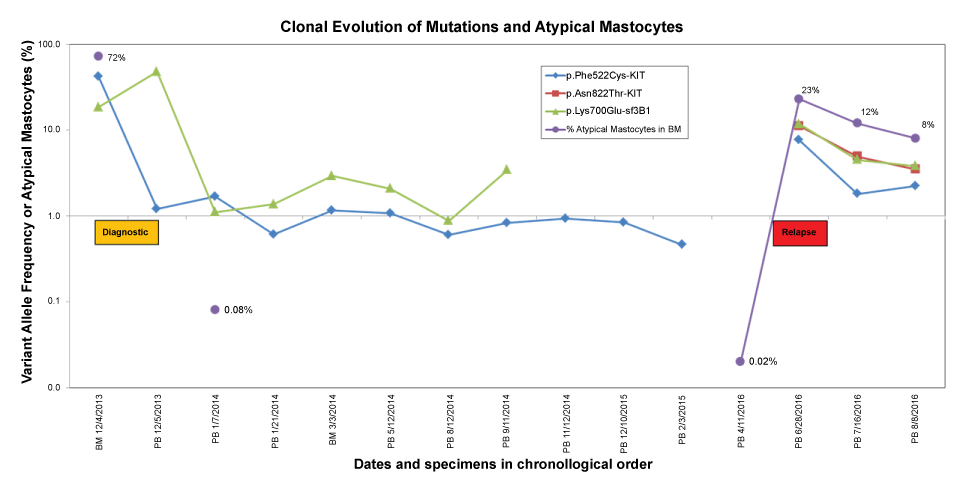 Figure 1: Time course of the clonal evolution of mutations in mastocytosis. Diagnostic (yellow box) and relapse (red box) specimens were collected during the times indicated. Values on the Y-axis are shown on a logarithmic scale. The percentage of atypical cells was measured by flow cytometry and it was below limit of detection during the complete remission time (< 0.002%). BM: Bone Marrow; PB: Peripheral Blood.
View Figure 1
Figure 1: Time course of the clonal evolution of mutations in mastocytosis. Diagnostic (yellow box) and relapse (red box) specimens were collected during the times indicated. Values on the Y-axis are shown on a logarithmic scale. The percentage of atypical cells was measured by flow cytometry and it was below limit of detection during the complete remission time (< 0.002%). BM: Bone Marrow; PB: Peripheral Blood.
View Figure 1
During disease progression, we found no mutations in other genes associated with mastocytosis (ETNK1, EZH2, STAT5A) [18] or any other known KIT mutations, based on a pediatric review of 1747 cases [19]. Moreover, we did not find a polymorphism that resulted in the amino acid substitution, p.Met541Leu [5,6,20,21].
The relevance of this case rests in the rareness of the acute form of MCL. To our knowledge, the present case study is the longest reported complete remission for a patient with MCL. Our findings highlighted the importance of sequencing the complete KIT gene to ensure the appropriate tyrosine-kinase inhibitor is selected, based on the detected KIT mutations. Our detection of the imatinib-sensitive p.Phe522Cys mutation in the KIT gene was important in selecting the correct treatment. This mutation only represents 5% of the total number of mutations associated with SM.
On the other hand, p.Lys700Glu is the most frequent SF3B1 mutation among hematological diseases which is associated with an abnormal selection of the 3' splice sites for a group of transcripts, including the iron transporter ABCB7, leading to a refractory anemia with ringed sideroblasts [22-24]. This mutation is very infrequent in SM and is associated with the ring sideroblast phenotype [7].
To understand the sensitivity to imatinib, the structures of KIT protein were analyzed at the atomic level. The autoinhibited conformation of KIT was observed in the tertiary structure of KIT bound to imatinib (PDB ID: 1T46) [25]. This conformation was previously resolved for ABL1 bound to imatinib fifteen years ago, by Nagar, et al. [26]. They showed that a mainly unstructured region, Arg815-Leu831, comprised the A-loop, which changed its conformation upon imatinib binding to the ATP binding site. In the inactive state, the side chains of two amino acids, Asp816 and Asn819, formed a hydrogen bond, which stabilized the conformation. This hydrogen bond could not form in the active form of KIT. In addition, Asp816 is an important amino acid, because its side change also stabilized the helix, by forming a dipole with a single-turn of the 310 helix, Ile817-Asp820. When trans-autophosphorylation of KIT takes place, the A-Loop is exposed to the solvent, and the guanidinium group of Arg815 forms a hydrogen bond with the Tyr823 side chain, which stabilizes the active conformation of KIT [27-29] (Figure 2).
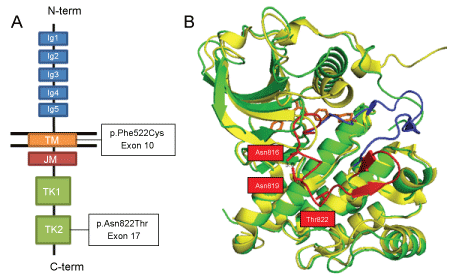 Figure 2: The KIT protein structure and functional mutations. A) Schematic representation of KIT domains and the locations of the two mutations found in the studied case; B) Tertiary structure of the inactivated KIT tyrosine-kinase domains, shown at the atomic level. The p.Asn822Thr mutant (PBD ID: 1T46_N822T; green) is overlaid onto the wild-type (PBD ID: 1PKG, yellow). The A-loops are depicted in red (inactive state) and blue (active state). The bound STI-571 (Imatinib) molecule is shown in orange.
View Figure 2
Figure 2: The KIT protein structure and functional mutations. A) Schematic representation of KIT domains and the locations of the two mutations found in the studied case; B) Tertiary structure of the inactivated KIT tyrosine-kinase domains, shown at the atomic level. The p.Asn822Thr mutant (PBD ID: 1T46_N822T; green) is overlaid onto the wild-type (PBD ID: 1PKG, yellow). The A-loops are depicted in red (inactive state) and blue (active state). The bound STI-571 (Imatinib) molecule is shown in orange.
View Figure 2
In our case, the mutant clone that appeared after the patient relapsed exhibited an alteration in the Asn822 residue. KIT mutations within the 822 codon were detected in a number of pediatric and adult patients with AML, sporadic and familial gastrointestinal stromal tumors, and seminomas. Although the vast majority of these mutations caused the p.Asn822Lys substitution, other KIT mutational substitutions were observed, including p.Asn822His [30,31], p.Asn822Thr, identified in a patient with pediatric AML, by Shimada, et al. [32] (as in our case), and p.Asn822Tyr, identified in a familial case of gastrointestinal stromal tumor [33].
The efficacy of imatinib binding to KIT proteins with Asn822 mutations remains unclear. Wang, et al. reported sensitivity to imatinib in leukemic cells with t(8;21) and p.Asn822Lys [34]. On the other hand, in vitro assays with Ba/F3 cells that expressed the KIT mutant p.Asn822Ile confirmed a resistance to imatinib [35]. A new clone appeared after relapse in our case. This double KIT mutant, p.Phe522Cys/p.Asn822Thr, which appeared after 2.5 years of 400 mg daily imatinib treatment, could have caused the patient's relapse. The relapse prompted us to change the tyrosine-kinase inhibitor to midostaurin, but the mutant was also resistant to this drug (Table 2).
Table 2: Effect of imatinib on mutations of KIT protein. View Table 2
We suspected that all pathogenic mutations found at relapse might have belonged to the same clone. In the case of the two mutations in the KIT gene, we could not demonstrate whether the two variants occurred in the same clone, due to technical limitations. The mutations were separated by 900 nucleotides, and our NGS reads covered 150 base pairs.
The Tyr823 residue plays a key role in the A-loop. It becomes phosphorylated in vivo, and due to the importance of the A-loop in the dysfunction of KIT, perhaps the normal operation of the protein was altered with the presence of the new, phosphorylatable hydroxyl group in the substituted Thr822 amino acid. Furthermore, the introduction of new putative hydrogen bonds might promote an aberrant, constitutive, stem cell factor-independent activation of the KIT protein.
Despite the presence of this mutation and the aberrant karyotype found in our patient, usually associated with MDS, the absence of ringed sideroblasts together with the rest of the findings associated with SM, made us think that we were facing a true cell leukemia mast cells. The mutation in SF3B1 served to monitor the patient's response, along with the KIT mutation. Subsequently, from the relapse, the similarity of the VAF data between the relapse mutation and that of SF3B1 could indicate that this could be the emerging clone that gave rise to the p.Thr822Asn KIT mutation (Table 1).
In conclusion, MCL is a rare disease; a correct diagnosis requires a combination of clinical findings, laboratory results, and imaging analyses. The molecular analysis of the KIT gene was crucial for selecting the appropriate treatment. A determination of the sensitivity of KIT to imatinib can provide a basis for remaining on alert for the emergence of future resistances. Further functional studies, including the KIT protein with double mutations, could lead to new insights that enhance our understanding of why new resistances appear.
These studies were conducted in accordance with the principles of the Declaration of Helsinki, and the protocols were approved by the appropriate institutional review boards. The patient provided written informed consent for the analysis of specimens. Informed consent for publication was obtained; it is available for review by the editor.
All authors declare no competing interests with respect to the research, authorship or publication of this article.
This work was supported by the Fundación CRIS and Red Temática de Investigación Cooperativa en Cáncer (RTICC); Instituto de Salud Carlos III. (Ref.: RD12/0036/0061, awarded to JML).
RS and JML contributed to the conception of the case report and designed and performed the molecular biology data analysis. RA, PM, TC, LL, and MLP contributed to patient care and the case presentation. RS, RA, and JML drafted and revised the manuscript. All authors read and approved the final manuscript.
Not applicable.