SARS-CoV-2, a new virus, which has been causing the catastrophic pandemic in the world in 2020, triggers numerous physiologic changes in humans, with potentially fatal evolution due to COVID-19. COVID-19 can trigger immunoparalysis with deep and silent immunosuppression and a state of tolerance that may elicit opportunities for neoplastic transformation or latent infectious diseases. In addition, all neurologic or psychiatric symptoms have been observed - as clinically present in usual diseases, but behind these phenomena, there is often the chronic phase of COVID-19 as a background, mediated by intense platelet activation. Symptoms may vary greatly as they depend on subjacent comorbidities but reflect a unique wide-spectrum syndrome ranging between absent or mild autoimmune undetermined disease and two classical diseases: Eosinophilic Granulomatosis with Polyangiitis (EGPA) and Granulomatosis with Polyangiitis (GPA). Interestingly, deviations in tryptophan catabolism cause neutrophilia with variable eosinophilia and apparently, tryptophan metabolism is altered both due to inflammation and niacin depletion. Lack of tryptophan causes sustained anaemia due to insufficient stimulation of the bone marrow. This article is based on articles reviewing, clinical observations, and high-intensity routine studies and in-hospital COVID-19 patients practice over 8 months. The main conclusion of this study is that the whole process starts among neutrophils and then is perpetuated by sustained platelet activation. Additional issues and hospital guides are explained in the Appendix. Appendix is recommended to fulfil the comprehension of the ideas contained in this theory.
Data for this Review article were gathered searching the MEDLINE, PubMed, Google academic databases using the following descriptors: "immune response and tryptophan", "tolerance and tryptophan or Kynurenine", "angiogenesis and tolerance", "5-HT and angiogenesis", "COVID-19 and NET", "COVID-19 and autoimmunity","COVID-19 pathophysiology","SARS-CoV-2 immune response", "SARS-CoV-2 immunosuppression". For the search without words "COVID-19" articles were selected in the period between 01/01/2000, given the research on themes as the tryptophan metabolic pathways exist before the "COVID-19 era". For articles containing the term "COVID-19", articles were chosen from November 2019 to December 2020. Only articles written in English were selected, preferably on high quality and research well-known publications.
COVID-19, Sars-cov-2 infection, Immunotolerance, Immunoparalysis, Hypereosinophilic syndromes, Tryptophan, NAD/NADH+
Throughout 2020, we have been facing the SARS-CoV-2 pandemic, and it is necessary to understand that COVID-19 is not a pulmonary disease only, but a systemic condition. SARS-CoV-2 main binding point, the Angiotensin Converting Enzyme-2 (ACE-2) receptor is overspread along many tissues throughout human body: Oral and nasal mucosae, gastric, gallbladder, bowel and colonic mucosae, urogenital tract including male gonads, eyes, vascular endothelia, heart muscle, kidney, and liver [1-3]. Based on this fact, a wider spectrum of clinical presentations and diagnoses can be expected, explaining clinical cases that could have underdiagnosed COVID-19-explained symptoms. Recently, studies and hypotheses have been pointing to unexpected and recurrent neurological symptoms in COVID-19 patients - sometimes not only by SARS-CoV-2 infection but also the latter associated with other viruses that show a central nervous system (CNS) tropism [2,4,5], which showed a negative swab PCR test obtained either from airway or cerebrospinal fluid (CSF). Not uncommonly, patients were admitted to hospital with new psychiatric presentations, showing no pulmonary symptoms [6,7], and those have evolved with respiratory decompensation within three or four days of hospitalization. Many of them were inappropriately diagnosed as nosocomial pneumonias - when should have been Covid-19 diagnosed should chest computer tomography (CT) scan have been performed, even by hospital admission moment. Yet, it remains vital to understand the ways SARS-CoV-2 affects our physiologic state as a systemic condition.
1) This study was performed at Hospital do Servidor Público Estadual (HSPE), a 794 beds general hospital facility built-in 1961 to assist civil public servants of São Paulo state, in Brazil. Adaptations to cope with the epidemic were performed in hospital structure during March 2020 - 307 beds had been intended to Covid-19 patients' treatment, 80 of which are intense care units (ICU) beds. We have been working hard among a multidisciplinary team: Physicians, nurses, and a range of professionals struggling to carry on. The Covid-19 cases admissions and caring have been performed under the Hospital's Risk Committee directors' supervision and infectious diseases management with all the involved medical specialities supporting this international emergency. 2,500 Covid-19 patients were admitted from March to October, and then we were, clearly confronting the second facet of this terrible story: There are the acute Covid-19 when there are circulant active viruses. Furthermore, patients admitted with inflammatory syndromes after being Covid-19 sick did not always recognise this condition, leading patients to die without the right diagnosis.
2) The infectious diseases speciality crew experienced an intense routine change during the pandemic, evaluating patients who were originally admitted for apparently not related problems, under the care of other medical experts and specialities. After admission, they presented positive markers or serologic tests for Covid-19 or even showed new suggestive symptoms. Most of these patients have been previously infected, and by then they showed inflammatory or immunosuppression states with periodic inflamed intervals, that have been erroneously diagnosed as acute Covid-19.
3) Still, during the first month of the pandemic in our country, a pattern was perceived among those patients who showed severe evolution, including our Hospital's patients. Severe cases occurred more often in elderly, diabetic, and obese patients- and the association could be noticed both in patients' clinical profiles and in laboratory findings.
4) We have been observing prevalent Hypocalcaemia among COVID-19 patients, along with other electrolyte alterations. Calcium (Ca2+) is known for cyclic fluctuation among blood cells components and takes part in symptoms often referred to. Our explaining hypothesis starts from this observation (Appendix).
5) For best discussing clinical presentation and treatment, we divided Covid-19 related inflammation into two categories:
1) Acute Covid-19 (the observed cytokine storm in early lung disease) and 2) Subacute and Chronic Covid-19 (the hyperinflammatory syndrome due to Covid-19). Based on these, a new treatment protocol based on a methylprednisolone pulse therapy was implemented between May and June 2020 (Figure 1, Figure 2 and Appendix) [8].
 Figure 1: The pulse of Methylprednisolone in acute COVID-19. A 45-years-old man with previous obesity, hypertension and diabetes mellitus type II. He is admitted to E.R. after six days after that symptoms have started. Referred had a fever, myalgia, and malaise. He was pulsed with Methylprednisolone 1000 mg for- 3 days, beginning - on the first day of admission, due to higher ferritin levels. He was discharged after 8 days. Follow up was performed. He returned to outpatient follow-up with myalgia and malaise. Exams were showed no discrepancies, but the CBC hemogram showed mild leukopenia and ferritin level was below the average reference value. He was prescribed B complex vitamin for 1 month and Prednisolone 60 mg/day for 5 days followed by 20 mg for more 5 days. The patient had an excellent clinical response. New follow-up is going to happen soon. (CBC: Complete blood count).
View Figure 1
Figure 1: The pulse of Methylprednisolone in acute COVID-19. A 45-years-old man with previous obesity, hypertension and diabetes mellitus type II. He is admitted to E.R. after six days after that symptoms have started. Referred had a fever, myalgia, and malaise. He was pulsed with Methylprednisolone 1000 mg for- 3 days, beginning - on the first day of admission, due to higher ferritin levels. He was discharged after 8 days. Follow up was performed. He returned to outpatient follow-up with myalgia and malaise. Exams were showed no discrepancies, but the CBC hemogram showed mild leukopenia and ferritin level was below the average reference value. He was prescribed B complex vitamin for 1 month and Prednisolone 60 mg/day for 5 days followed by 20 mg for more 5 days. The patient had an excellent clinical response. New follow-up is going to happen soon. (CBC: Complete blood count).
View Figure 1
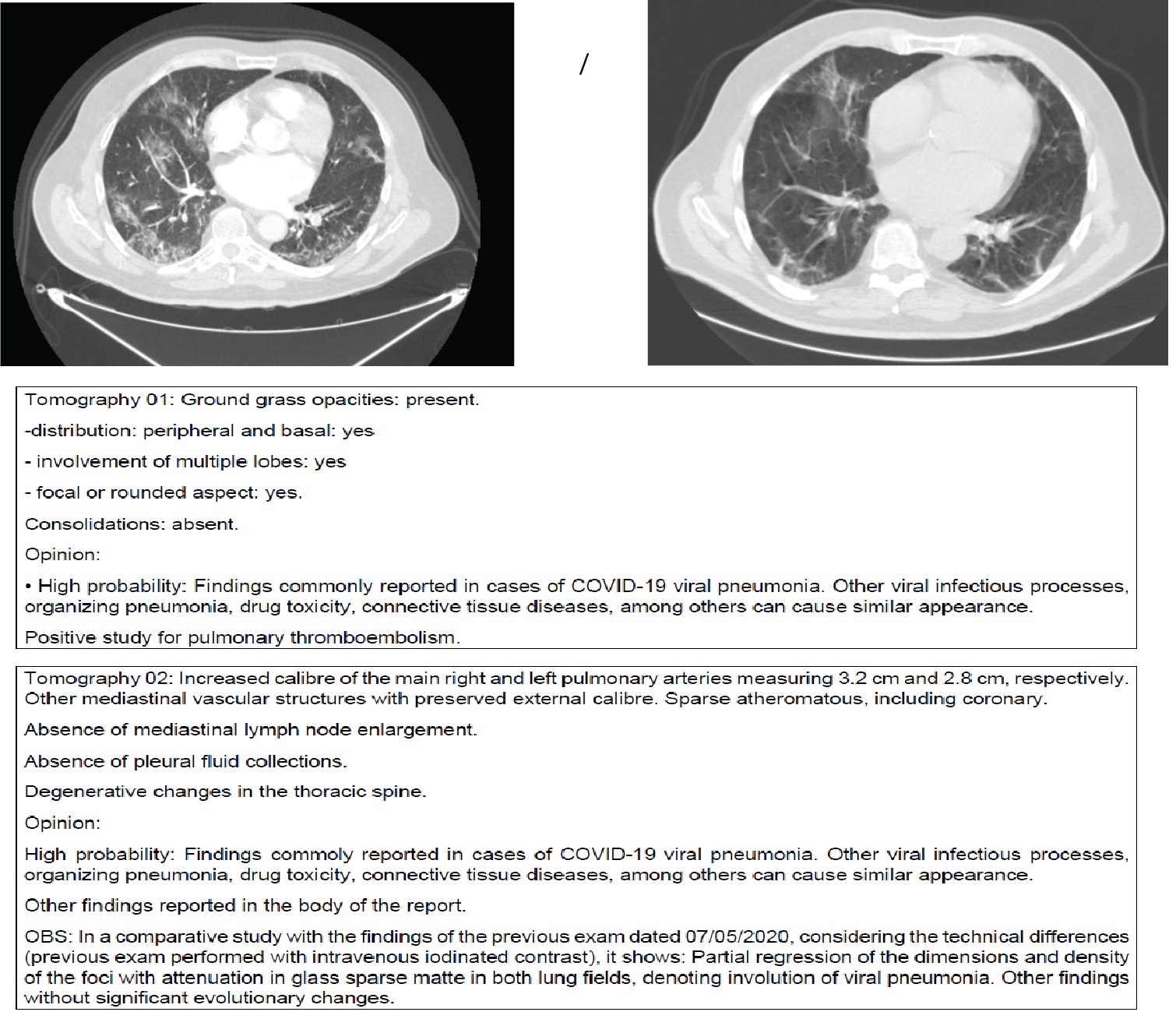 Figure 2: The pulse of Methylprednisolone in chronic COVID-19. Patient, 68-years-old, previously admitted due to COVID-19.
Figure 2: The pulse of Methylprednisolone in chronic COVID-19. Patient, 68-years-old, previously admitted due to COVID-19.
After 7 days of hospital discharge, he had worsening effects in-breath and systemic symptoms such as fever and malaise.
He was pulsed with Methylprednisolone 1g 3 days plus 5 days with 80 mg (first protocol). Exams not shown except for
Urine with Enterococcus faecalis ampicillin SENSITIVE. He had improved and was discharged 1 day after the pulse (9 days
after) with follow-up. A problem that is happening is recognizing these patients as being in the acute phase or community
pneumonia. Acute phase has soft image at tomography, and chronic COVID-19 with worsening conditions in breath has
more response to corticosteroids in low doses and less time of orotracheal intubation.
View Figure 2
6) This article intends to elucidate Covid-19 complex metabolic alterations, reviewing biochemical cycles and summaries to understand the disease and its impacts better. It also means to show that considering chronic Covid-19 as an acute disease is especially problematic: generating familiar and social distancing and performing incorrect treatment due to a misunderstanding of this new phase of the disease's physiopathology. Covid-19 also often mimics surgical conditions and affects the postsurgical recovery. It has been causing clinical recurrences and re-hospitalisations for not-related to Covid-19 conditions.
7) This article brings some theoretical pathophysiological explanations for the observed phenomena, based both on literature reviews and clinical observation. It is well-known that randomised clinical trials consist of the best evidence for treatment guidelines - but those trials demand time to be performed. There has been some difficulty dealing with public demand for a treatment guideline, but it is necessary to understand how this new condition causes changes to our bodies before we can achieve definite guideline or best classifications. These demands more than usual protocols to acquire technical knowledge for a best-defined treatment or risk predictor.
Our theory divides Covid-19 into two phases: Early (the first 14 days of infection) and chronic. Early phase: The most remarkable changes noticed in this phase is a viral activity in the lungs, but other ACE-2 expressing organs also take part, and many different symptoms may occur. In this phase and to classic symptoms, we must recognise an ischemic stroke, acute myocardial infarct, behavioural changes, consciousness fluctuations and other phenomena as early Covid-19 symptoms. Ischemic events may be due to vasospasm and thrombosis; meningoencephalitis may be due to herpes reactivation [1,2,9-14].
Trying to understand Covid-19's physiopathology has not been simple - yet it seems to be even more complex than it looked. Based on the first studies, published before the disease has reached our country, we have proposed theories and explanations about SARS-CoV-2 infection and guided treatments based on ACE-2 and RASS disturbances. It became clear that viral actions have important roles in Covid-19 signs and symptoms through clinical observations. Nevertheless, it was especially intriguing why obese, diabetic, or elderly patients have more severe disease, whether or not having previous cardiac conditions.
Another early-noted symptom was some reactive arthritis. These initial findings together led to two complementary theories.
SARS-CoV-2 direct actions, immune response and the origin of autoantibodies and acute ischaemic phenomena: Opening doors for autoimmunity.
From the first observations, it became clear that children and adults may develop Covid-19: But the experience the same disease in different spectra: Adults have been presenting some catastrophic thrombotic events and another myriad of symptoms usually seen in autoimmune diseases. Our hypothesis lays in an immune imbalance leading to severe disease (Figure 3).
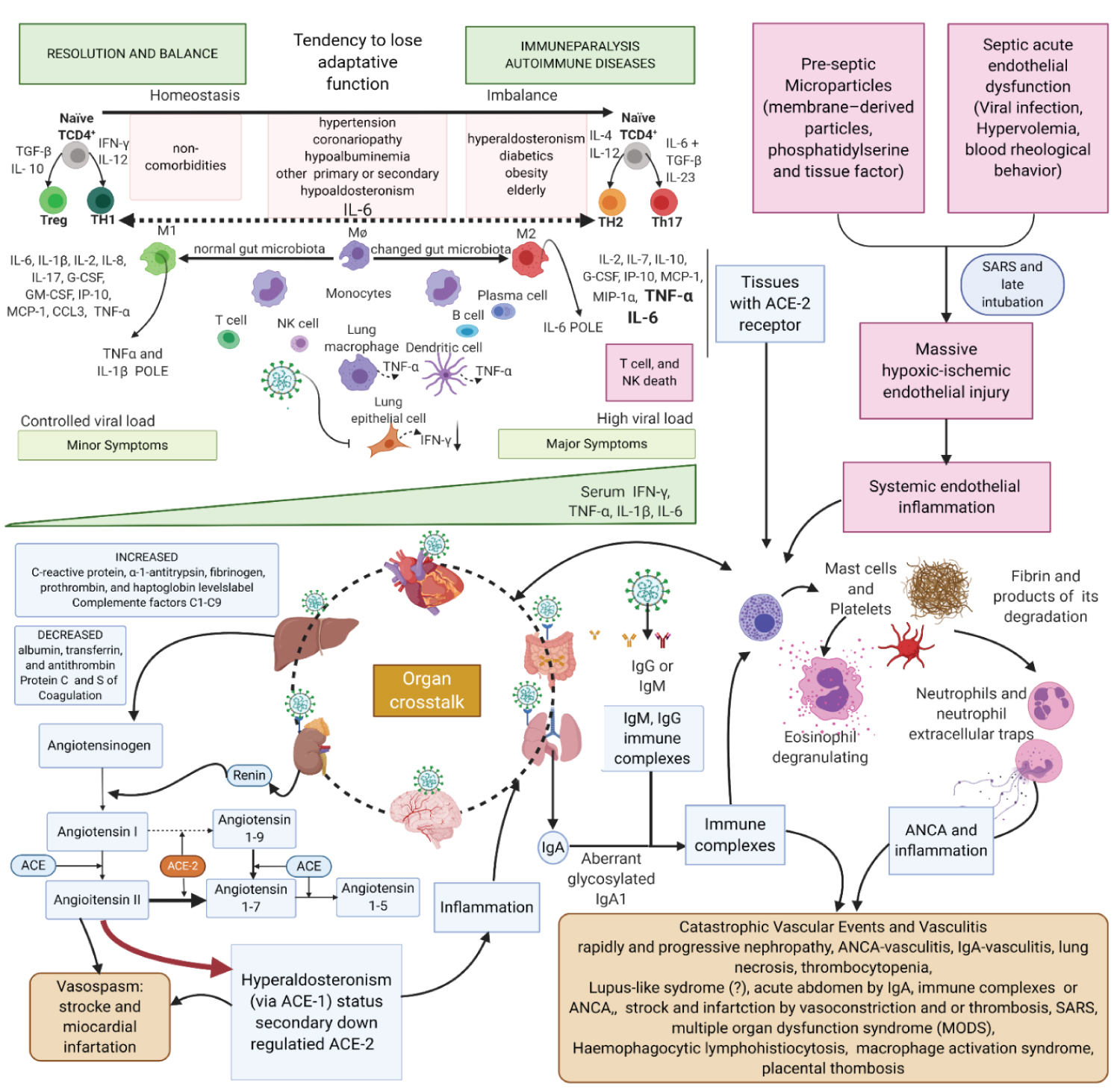 Figure 3: Created with BioRender.com
Figure 3: Created with BioRender.com
First Theory. Ischaemic mechanisms in COVID-19 physiopathology. Left upper Figure: Patients under certain conditions (elderly, obese, diabetic) have the innate immunity pathway more activated than adaptive immunity. Elderly, due to immunosenescence, have a natural decrease in TCR (T cell receptor) and B cells; obese patients show has activation of M2 macrophages M2 (anti-inflammatory) in to the detriment of M1 macrophages M1 (inflammatory) to balance inflammatory characteristics of comorbidity. Diabetic patients present glycosylation of the inflammatory pathways causing decrease in the adaptive immune response and an increase in the innate one. Some studies have already shown lasting IgA in high concentration in sera compared to IgM and IgG. IgA1 may be glycosylated differently, triggering production of autoantibodies forming immune complexes when they bind to serum IgA1. Right upper Figure: Sustained hypoxemic status and microparticles (MPs) also contribute to the worsening conditions in the endothelial inflammation. MPs are cell membrane-derived particles that can promote coagulation, inflammation, and angiogenesis and can participate in cell-to-cell communication. Sometimes late intubation can worsen existing inflammation. Lower Figure: Immune complexes are also formed in the opsonization process by IgM and IgG. Immune complexes start the cascade of kidney damage. IgA nephropathy has haematuria and proteinuria that can be massive in a stage of nephrotic syndrome. Erythrocytes are injured in the glomeruli and release chemotactic contents to the complement system, which increase the renal inflammatory response. IgA causes vasculitis and may explain Kawasaki syndrome in children, thrombotic events, and abdominal tenderness in adult patients. Hypercoagulative status is intense due to inflammation and increased production of procoagulant factors, such as fibrinogen, produced by the liver; or by the loss of anticoagulant elements, like protein S and protein C lost in nephrotic syndrome. ACE-2 receptor is down- regulated in the tissues due to infection by SARS-CoV-2; thus, the anti-inflammatory and vasodilatory pathway is impaired, diverting Renin-Angiotensin-Aldosterone System (RAAS) to aldosterone. In the kidneys, these effects with hyperactivated macrophages lead to a rapidly and progressive renal failure, as seen in severe cases. Serum immune complexes and their deposition in tissues activate complement system that induces mast cells degranulation (causing urticaria), attracting neutrophils to the inflamed tissue. Some studies have shown the action of neutrophil extracellular traps (NETs) on SARS-CoV-2 infection. NETs play an important role in the evolution of COVID-19 to severe forms, perhaps stimulating tissue necrosis, increasing local inflammation, leading the patient to present Severe Acute Respiratory Syndrome (SARS). NETs have the function of helping the organism in the control against infections, it seems to have an intensifying effect on B cells and act, in some way, in the polarization to TCD4+ of adaptive immunity. NETs are also related to the formation of antineutrophil cytoplasmic antibody (ANCA), which can cause vasculitis. Organ crosstalk is a mutual communication between distant organs to maintain the homeostasis. ACE-2 receptor is prevalent in human body and the inflammatory syndrome is not a unique organ syndrome, it is an important and intense talk between organs that explains the massive inflammatory response in COVID-19.
View Figure 3
At the initial phase of the disease, macrophage TNF-α production is inhibited, and dendritic cells have decreased HLA class II expression by SARS-CoV-2 infection. The adaptive immune pathway shows a weak T cell response to eliminate the virus due to ageing, obesity, or diabetes mellitus, and these patients have high viral replication rates and antibody release by the innate response and immune complex formation [15,16]. Long-lasting immune complexes (IgA, IgM, IgG) induce type III hypersensitive reactions in subacute and late phases, activating the complement system and mast cell degranulation with platelet aggregation in different tissues. For this reason, neutrophilia and elevated inflammatory markers "de novo" are not uncommon: In response to accelerated viral replication and viral particle overspread in tissues with the ACE-2 receptor, macrophages are no longer inhibited, and monocyte-derived macrophages chemoattract neutrophils.
Neutrophils are bacterial killer specialized cells, sometimes by neutrophil extracellular trap (NET) formation (NETosis), whose role in viral infections is unknown [1-30]. NETs cause IL-6-mediated cytokine storms; furthermore, NETs are antineutrophil cytoplasmic antibody (ANCA) inducers that can contribute to vasculitis. SARS-CoV-2 infection-induced autoantibodies trigger different symptoms.
Multiple inflammation pathways combining immune complexes, direct viral damage, NET formation, proinflammatory aldosterone action and disorganised macrophage activation leading to hemophagocytic lymph histiocytosis are critical elements of thrombosis background phenomena understanding. This hypothesis may explain the rapid and progressive kidney injury, increasing proteinuria and haematuria, which we have considered a predictor of COVID-19 severity, as we have been assessing in our practice. In the intensive care unit, most severe Covid-19 patients show proteinuria (+++/3+) and haematuria, and sometimes unavoidable haemodialysis need. We consider urine a marker to interrupt anticoagulation and corticosteroid use [4].
The second phase begins nearly seven days after the appearance of the first symptoms. It can be defined as a hyperinflammatory syndrome: An unknown and unrecognised disease by most physicians - and denied by others, who consider this late inflammation period secondary to new bacterial infection and sepsis. This phenomenon has represented a significant cause of death in chronic COVID-19 infection.
After 14 days, patients - whether adults or children - are at risk of dying from COVID-19 due to an inflammatory syndrome, because the same virus infects both groups. SARS-CoV-2 and ACE-2 receptors/coreceptors sustain the main physiopathology that explains acute symptoms and chronic inflammation's persistence with a plethora of symptoms (Figure 4a-c). ACE-2 is internalised and downregulated by SARS-CoV-2 infection, causing changes in RASS. This reinforces inflammation and promotes low tryptophan (Trp) absorption, which may produce its acute depletion [31-35]. Trp is an essential amino acid that related to immune response polarisation either to tolerance or to inflammation. ACE-2 mediates tryptophan absorption in enterocytes due to B0AT1 expression, a sodium-dependent neutral amino acid transporter found in the small bowel epithelium. Without proper transport molecules to allow tryptophan to enter the enterocytes and consequent Trp starvation, dangerous consequences begin [35-37].
 Figure 4a: Created with BioRender.com
Figure 4a: Created with BioRender.com
Genesis and Apocalypse together: apoptosis and death; neovasogenesis and tumorigenesis. IL-6, TNF-α, INF-γ variation during the natural history of COVID-19. It is essential that, in the acute phase of the disease, there is the presence of viruses in a non-immunosuppressed patient until about six days after the onset of symptoms. In chronic COVID-19, the patient can be admitted either due to a new infection by SARS-CoV-2, as this patient is in immunoparalysis, or is not in a new infection, but he had to worsen of the inflammatory disease triggered by SARS-CoV-2, causing the state of tolerance. The hypothesis is that SARS-CoV-2 generates IgG-containing immune complexes by the hosts and an intense migration of neutrophil due to IL-6 polarization. Haematopoietic cells, which express three classes of Fc receptors (FcγRI, -II, -III), can activate neutrophils and monocytes by interacting with immune complexes. Neutrophil stimulation with cytokines (such as C5a or TNF-α) and ANCA results in respiratory burst and degranulation, which leads to the release of tissue factor (TF)-bearing microparticles and NETs that subsequently activate the coagulation system and generate thrombin. This leads to the generation of more C5a, establishing a self-fuelling inflammatory amplification loop leading to the vasculitis injury. Endothelial nitric oxide synthase (eNOS) has a central role in vascular function and angiogenesis and vascular endothelial growth factor (VEGF) and an increase in intracellular calcium concentration stimulate eNOS activation in healthy endothelial cells. eNOS also induces telomerase activity, and mitochondrial biogenesis and hypoxemia stop this factor of protection. Massive endothelial injury is a cause of maintenance of continuous injury. Neutrophil firm adhesion transmits signals to the endothelium through β2-integrins/ICAM-1 interactions, leading to a rise of cytosolic free Ca2+ and cytoskeletal changes required for neutrophil diapedesis. This signalling produces a microvascular permeability, which is further enhanced by proteases and oxidants released at the immediate proximity of endothelial cells during TNF-induced adhesion in the presence of ANCA. Endothelial cell detachment results in denudation of the subendothelial matrix leading to fibrin deposition, which implies platelet recruitment and thrombosis. It is a glycoprotein (GP) that shares significant amino acid sequence homology with erythropoietin (EPO). Thrombopoietin (TPO) is made in the liver by both parenchymal cells and sinusoidal endothelial cells and is secreted into the circulation at a constant rate. TPO bindings to the surface of platelets and megakaryocytes through the c-mpl receptor. Stimulation of the TPO receptor results in activation of Janus kinase type 2 (JAK2) and tyrosine kinase 2 (TYK2) that is important for cytokines that use the gp130 receptor and for some IFNs and is also important for the hormone-like cytokines such as growth hormone (GH), prolactin (PRL), erythropoietin (EPO), thrombopoietin (TPO) and the family of cytokines that signal through the IL-3 receptor (IL-3, IL-5 and granulocyte-macrophage colony-stimulating factor, GM-CSF). Hypoxemia induces a tolerogenic and tumorigenic environment.
View Figure 4a
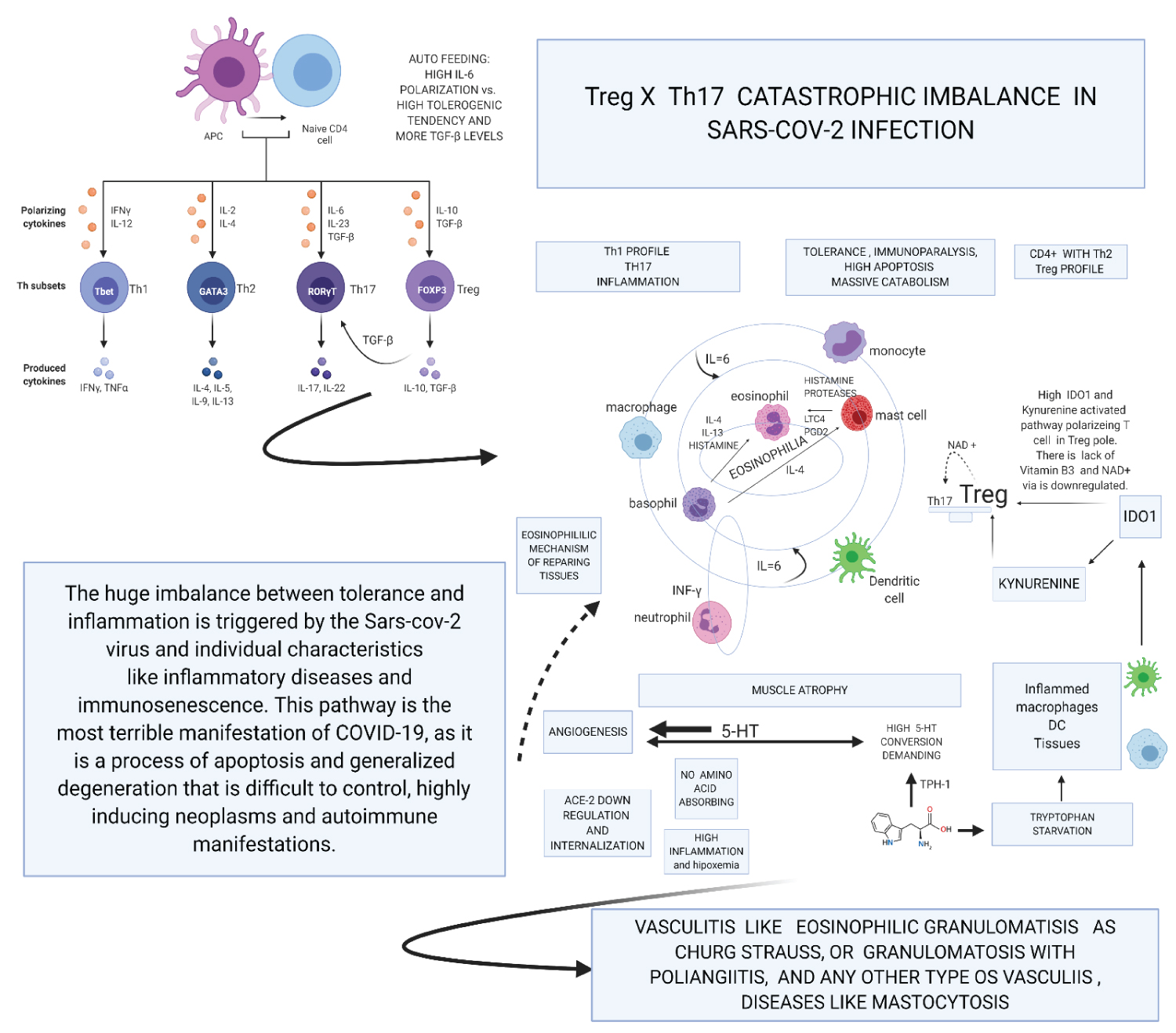 Figure 4b: Created with BioRender.com
Figure 4b: Created with BioRender.com
Second Theory. Catastrophic Treg x T17 imbalance in SARS-CoV-2 infection and Tryptophan starvation: SARS-CoV-2 infection by ACE-2 internalization and virus capacity of inhibiting. SARS-CoV-2 infection occurs internalization of ACE-2 and inhibition of INF-gamma by viral action. These are the first forms of polarization for IL-6, diabetic, obese, and older adults and other diseases present this polarization more accentuated because their comorbidities frequently inflame them. SARS-CoV-2 is a disease that promotes angiogenesis and great tolerance due to the TH17 and Treg imbalance.
On the one hand, we have the infection promoting a tendency towards TH1, but inhibited by INF-gamma regulation. The lymphocytes tend towards Treg and this TCD4 + Th2 function establishes a negative relationship in the control of the Th1 pathway, thus NK, Th1, TCD8 + initiate a process of apoptosis and downregulation of its active pathways in inflammation. The large production of ADO1 accentuates this pathway by macrophages and dendritic cells, so the Tryptophan pathway is aimed at inhibition, that is, tolerance. The high consumption of 5-HT by the angiogenic process depletes serotonin, bypassing the tryptophan pathway to produce this essential molecule in angiogenesis and in the development of tumours it is a vasodilator stimulus. Lack of Tryptophan in the tolerogenic Indoleamine 2,3-dioxigenase (ADO)1 pathway polarizes immune system to Treg. The great lack of tryptophan promotes the consumptive syndrome by sarcopenia and the lack of vitamin B3, shifting the metabolism towards anaerobic respiration due to the lack of NAD/NADH+. NAD+ has a positive role in polarizing Treg to Th17, but it is made impossible by the anaerobic pathway. This process together triggers a metabolic explosion with high apoptosis that is being self-fuelled by the production of IL-6 via Th2 and by monocytes and macrophages stimulated continuously by new tissue lesions or new bacterial infections, mainly by gram negatives that stimulate IFN-γ. In this hurricane, even more, exacerbated, the constant pro-inflammatory stimulus sustains an established tolerogenic state.
Thus, constant inflammation vis-a-vis a state of tolerance; there is a high probability of autoimmune diseases. Also, the toxic metabolites of tryptophan produce many neurological symptoms. Concerning the vessels, the continuous process of aggregation and reconstruction facilitates thrombosis mechanisms, and the high expression of adhesins associated with the demand for 5-HT is highly chemotactic for eosinophils. Eosinophils pass through all tissues so that a large systemic hypereosinophilic syndrome mediated by serotonin is present, IL-6 activating mast cells and basophils. Finally, all types of eosinophilic vasculitis can happen in these patients. The intensity of all this complex pathophysiology depends on the inflammatory diseases that the patient has as comorbidities and on the vascular injury caused by the viral infection, so one should never avoid intubation in a patient who is on the threshold of the PaO2/FiO2 ratio. Not intubating is damaging more endothelium and pushing the metabolic and immune uncontrolled explained here. The centre of the Figure shows that perhaps some eosinophilia or a lot of tissue eosinophilia depending on how the interaction between cells and cytokines occurs. It is important to note the different orbit of neutrophils that are responsible, above all, for the formation of NETs and are very importantly linked to platelet activation. Serotonin (5-HT) neutrophils follow an orbit and separate, as their stimulus, in this pathophysiology, is mainly influenced by autoantibodies of the ANCA type, although others may also appear and be involved. Even so, the design in the form of orbits intends to show that all polymorphs influence each other depending on the distance and the alignment between them. This characteristic, together with the specificities of each patient and hypoxemia, will be the conductor which orchestrates the signs and symptoms presented by the patient. The cause of variable eosinophilia and the unique orbit of neutrophils appears to be linked to the problematic metabolism of tryptophan, whose shift to Kynurenine may be the cause of cyclic neutrophilia (Appendix).
View Figure 4b
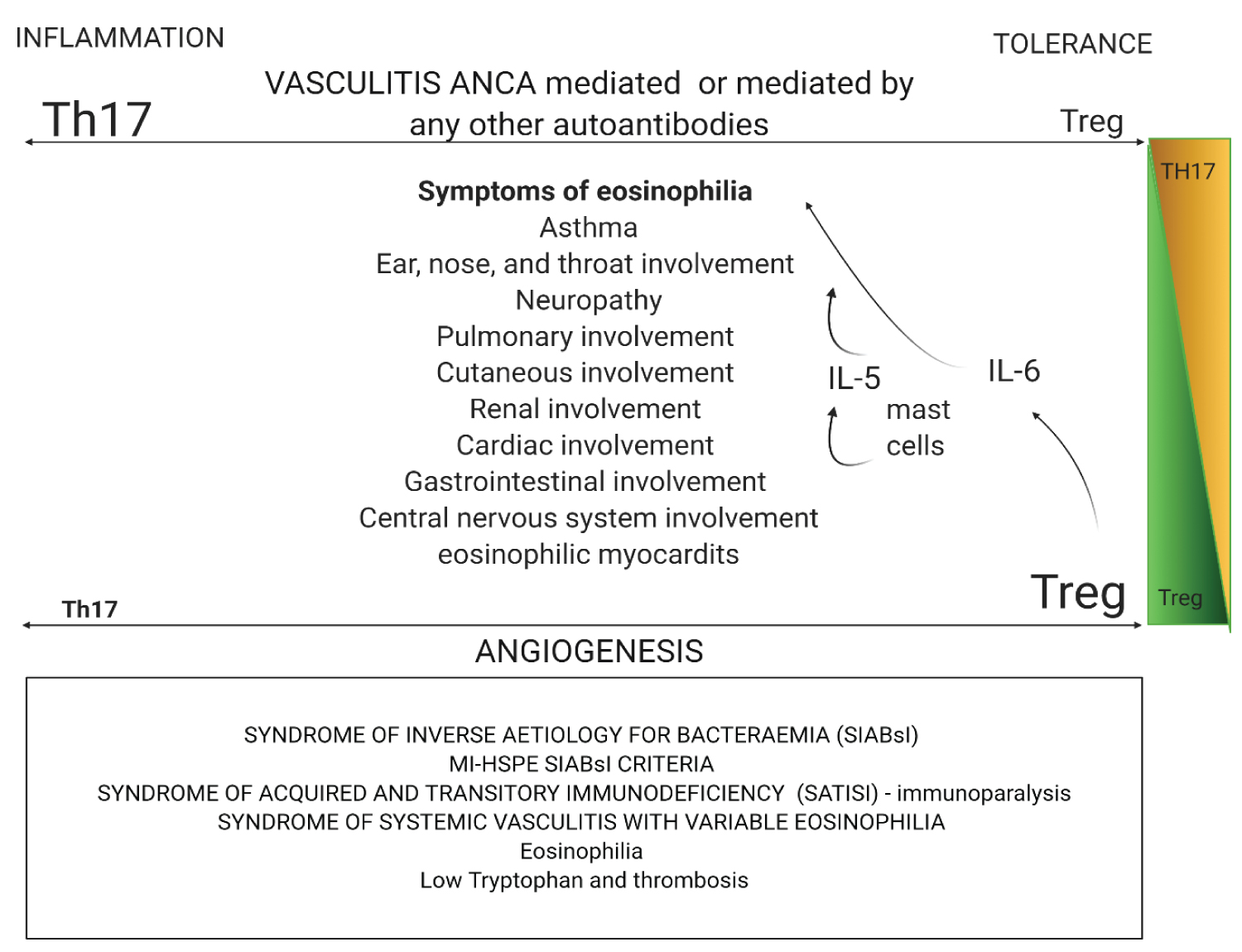 Figure 4c: Created with BioRender.com
Figure 4c: Created with BioRender.com
Eosinophilia between Genesis and Apocalypse: Eosinophilia between Genesis and Apocalypse: Eosinophilic granulomatosis with polyangiitis (EGPA) linked the dysregulation of PR3 because it is the main target of c-ANCA. PR3 is a cationic protein that consists of 228 amino acid residues and belongs to the serine protease family of trypsins. This imbalance of immune pathways is accompanied by the activation of plasmacytoid dendritic cells and T-helper-2 (Th2), Th9 and Th17 cells' polarisation. Polarization is for Th17 in EGPA. Resulting in circulating eosinophils that interact with endothelial cells, then migrate through the vessel wall and, finally, infiltrate a target organ through a regulated process involving the interaction between adhesion molecules, chemokine receptors in eosinophils (CCR3). IL-6 activates mast cells, and mast cells release IL-5, which is an eosinophilic chemo attractor. These specific granules for eosinophils contain cationic proteins, including primary base protein, eosinophil cationic protein, eosinophil peroxidase and eosinophil-derived neurotoxin. Eosinophils are responsible for tissue repairing and systemic symptoms in EGPA. This figure also shows syndromes that appear in COVID-19 during IL-6 immunoparalysis, or due to IL-6, Treg and Tryptophan influences.
View Figure 4c
For further information on our clinical practice-based hypotheses, please refer to Appendix, where all of them are enrolled in chronologic sequence.
Clinical practice in HSPE has been an excellent opportunity to treat and carefully observe numerous Covid-19 hospitalised patients, as long as their laboratory results and findings. In the first weeks of the pandemic, we had some hard time trying to manage recently ICU discharged patients - they showed a disproportional consumptive syndrome, unexplained only by hospitalisation staying time or catecholamines used to promote vasoconstriction. Based on a German hospital experience, we have settled a protocol for laboratory exams to be collected at admission time and periodic reassessment. This was crucial for observing the importance of simple, basic lab tests assessment when facing an unknown condition. In our findings, simple urine samples may point to understanding the kidney aggression site - and that leads us to consider evolving proteinuria as a severity predictor.
We have to keep in mind that we are still facing a war against an unknown virus. We are building knowledge and are now miles away from where we started. Standard lung images obtained when patients with systemic symptoms were readmitted (sometimes repeatedly) to Hospital showed acute Covid-19 mimicking findings. Some of those images are attached in Figures 6a-l.
More surprisingly, we must understand Covid-19 as more than an infectious disease. It is an infectious, metabolic and deficiency disease, related to comorbidities especially those with inflammatory characters, previously present. It can evolve more severely in patients with inflammatory conditions than compared to malnourished patients, for example. Malnutrition can be corrected throughout hospitalisation stay, while the genetic settings leading to inflammatory conditions are much more challenging to manage.
We noted common severe disease predictors among those patients whose profiles were pronated to a worse evolution. They show an intracellular aerobic respiratory chain cycle that is completely shaken by lack of essential vitamin B3, which originated from the tryptophan cycle. This explains some studies findings for a positive outcome found in the replacement of complex B vitamins in critically ill patients, not found for other vitamins. Covid-19 patients are under high-intensity metabolic stress, with depleted NAD/NADH+ to connect aerobic metabolism to anaerobic respiration. Also, in this highly intense stress period, blood cortisol is found in high concentrations.
COVID-19 is a complex combination of viral action and metabolic deficits triggered by infection and internalisation of ACE-2, modifying the RASS axis to inflammatory vasospasm and thrombotic pathway, and reaches its maximum when immunoparalysis and tolerance allow the development of neoplasms, which the immune system should contain.
Basal IL-6 is already polarised in chronic inflammatory conditions. When there is increasing pressure to reassure this deviation, these patients develop massive and catastrophic immunosuppression. This can explain the reactivation of latent infectious diseases observed in Covid-19 patients.
In this context, an inflamed bowel represents a significant and continuous supplier for Gram-negative bacteria and enterococci, leading to prolonged hospital stays and several antimicrobials' cycles, which show no effect on these bacteria. This condition is expected, given the phagocytosis capacity loss by polymorphonuclear cells under a permanent inflammatory state combined with exposed intestinal mucosae.
In this phase, the observed neurological findings are bizarre and intriguing and have not been explained or accepted. Platelets secrete IL-6 by multiple mechanisms, stimulating mast cells and this cytokine is overexpressed in people who have chronic inflammatory diseases. This way, they have a previous environment. With Sars-CoV-2 infection, a recrudescence provided a more intense mast cell, basophils and finally eosinophils mainly by IL-5 chemoattraction. There is a tendency towards neutropenia developing. Nevertheless, intense recruitment by bacterial translocation, systemic endothelial aggregation (motivated by low cellular respiration), autoantibodies, the complement cascade, and the tryptophan pathway (Appendix) make neutrophilia common; however, this finding is related to a poorer prognosis. The chronic inflammatory process mimics a sinusoidal shaped curve, with inflammation peaks followed by a nadir with a decrease in inflammatory laboratory markers such as C-reactive protein, D-Dimer, oxaloacetic transaminase, and lactic dehydrogenase (DHL). Notwithstanding, after 5 to 7 days, there is a progressive neutrophilic march with repeating patterns: Worsening of laboratory markers, respiratory performance worsens, D-Dimer and HDL rise and new anguish permeates everyone who is caring for the patient. Many facts were associated with the severe form of the disease, and we can mention among them several genetic conditions (HLA-DR with high expression, for example), lifestyle and the environmental conditions, viral load to which it was exposed, the type of inflammatory comorbidity - with particular importance for fat metabolism and immunosenescence. Clinical experience has taught us that the first point to think about is that obese and diabetic patients should never have delayed intubation and that all intubation for this patient profile should be performed in a "golden time". Since the beginning of the pandemic, we have been performing it, and efforts were made to define the main predictors for orotracheal intubation. However, all results have shown very controversial information and based on the doubt of prolonging hypoxemia; we chose to perform intubation by rigorous criteria: Pulse oximetry oxygen saturation (SatO2) equals to 93% or less - with an oxygen offer of at most 50%; breathing rate (BR) = 30 inspirations per minute or more, dyspnoea, choppy speech, Glasgow Coma Scale score 8 points or less. Current studies have proven that ongoing hypoxemia is the major endothelium inflammation factor leading to a bad prognosis. The tryptophan pathway is impaired both by the infection and hypoxemia, and its consequences will rely on the individual Trp reserve and present comorbidities (Figure 5). When there is an intense gastrointestinal infection by SARS-CoV-2, Trp absorption is deficient - explained by internalisation and downregulation of ACE-2 by inflammation.
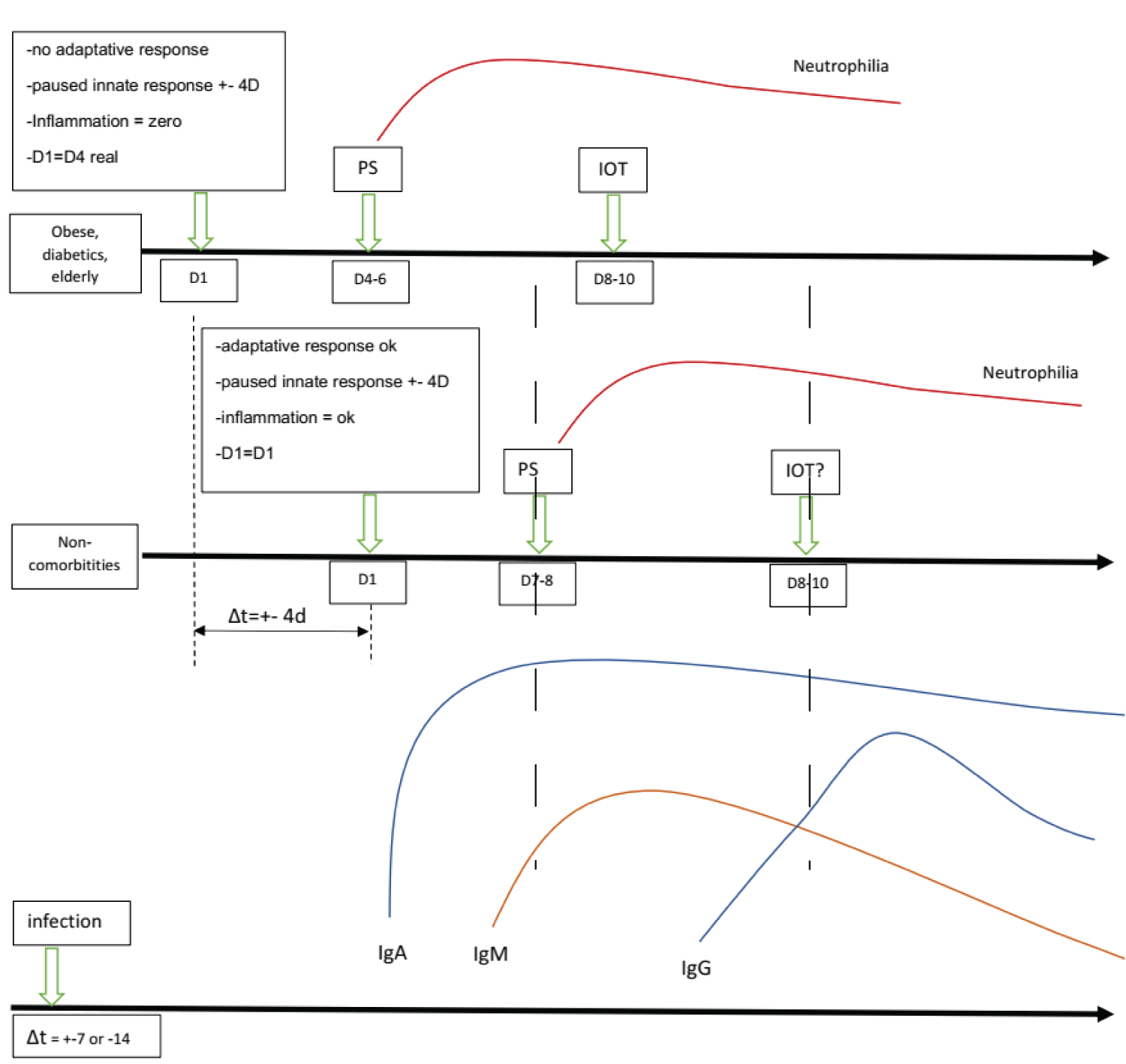 Figure 5: Paradoxical Theory "Back to the Present": The immune system is geared towards tolerance in people with inflammatory comorbidities and they will take a little longer to feel the effect of the inflammation or will present severe hypoxia when apparently, they seem well. Also, the elderly, diabetics and obese patients have a compromised adaptive system. When these patients are admitted reporting 3 or 4 days of symptoms, they have a chest tomography compatible with an evolution time of more than 7 days of illness. This population is highly vulnerable because of the lack of knowledge about the disease's pathophysiology has led these patients to have a delayed or not even performed intubation. SARS-CoV-2 pauses INF-γ at the beginning of de viraemic phase; D1 for comorbidities patients is actually D4 ( considering Tomography pattern); inflammatory comorbidities have a low inflammatory reaction due to paused INF-γ and poor adaptative response; these patients are in A SILENT VIREMIA and IN A SILENT HIPOXEMIA; Neutrophilic phase observed in a hemogram (pulmonary phase) appears in the same period in patients with inflammatory comorbidities and in non-inflammatory comorbidities, but what is shown is a delay of about 4 days between the two groups. D8 in comorbidities patients = D12 actually: we are not intubating these patients "in time." The virus tricked us. Intubating "in time" for comorbidities = intubating when patients arrive at thee.r if satO2 < 93%, since they are inwards about d7-d8 (when they are in D10-D12). The virus does not change the antibody kinetics changing our DNA in this case, but SARS-CoV-2 has mechanisms to polarize our immune system not to produce antibodies. PS = Emergency room, IOT = Orotracheal intubation, D4 = day 4 after symptoms.
View Figure 5
Figure 5: Paradoxical Theory "Back to the Present": The immune system is geared towards tolerance in people with inflammatory comorbidities and they will take a little longer to feel the effect of the inflammation or will present severe hypoxia when apparently, they seem well. Also, the elderly, diabetics and obese patients have a compromised adaptive system. When these patients are admitted reporting 3 or 4 days of symptoms, they have a chest tomography compatible with an evolution time of more than 7 days of illness. This population is highly vulnerable because of the lack of knowledge about the disease's pathophysiology has led these patients to have a delayed or not even performed intubation. SARS-CoV-2 pauses INF-γ at the beginning of de viraemic phase; D1 for comorbidities patients is actually D4 ( considering Tomography pattern); inflammatory comorbidities have a low inflammatory reaction due to paused INF-γ and poor adaptative response; these patients are in A SILENT VIREMIA and IN A SILENT HIPOXEMIA; Neutrophilic phase observed in a hemogram (pulmonary phase) appears in the same period in patients with inflammatory comorbidities and in non-inflammatory comorbidities, but what is shown is a delay of about 4 days between the two groups. D8 in comorbidities patients = D12 actually: we are not intubating these patients "in time." The virus tricked us. Intubating "in time" for comorbidities = intubating when patients arrive at thee.r if satO2 < 93%, since they are inwards about d7-d8 (when they are in D10-D12). The virus does not change the antibody kinetics changing our DNA in this case, but SARS-CoV-2 has mechanisms to polarize our immune system not to produce antibodies. PS = Emergency room, IOT = Orotracheal intubation, D4 = day 4 after symptoms.
View Figure 5
We believe that one of the most important pathways for thrombosis phenomenon physiopathology is that 5-hydroxytryptamine (5-HT) is present in the vascular endothelium since coating with 5-HT products prevents the expression of adhesins. In the absence of this protection, adhesins play an essential role in the coagulation cascade. These facts and the increase in tissue factor by activated monocytes and other polymorphonuclear cells are constant stimuli for thrombosis.
Also, changes in Trp cascades can elicit different bone marrow responses facing SARS-CoV-2 infection (Appendix).
The missing link for understanding and managing these outcomes seems to be eosinophilia. It explains a profile of the systemic disease, as we have found through clinical experience and daily managing of Covid-19 patients. Dermatologic findings, CNS symptoms, abdominal and cardiac alterations, worsening of respiratory parameters seem to be connected to eosinophilia (Figure 4c and Figure 6j-l). These patients show intense syndromic symptoms, with severe vasculitis and a malignant profile. The eosinophilia may not be perceived by blood count testing: eosinophils are attracted to blood vessels by cytokines and neutrophil released products, as long as activated mast cells, fibrin deposits and 5-HT due to coagulation cascades activation.
 Figure 6A-D: After ICU stay and orotracheal intubation and readmitted in the Infectious Diseases wards. Obese and or Diabetic patients after 14 days of disease. (14-40 days). Observe the "heavy" pattern of the images with opacities denoting extensive posterior atelectasis or even condensation in the pulmonary parenchyma in a diffuse manner. This tomography is different from the acute COVID-19 in Figure 1, where we have a ground grass pattern that is lighter and more tenuous. We are readmitting patients who already arrive with a "heavy" image pattern with new decompensation. This patient is not acute COVID-19; he is only exacerbating a disease, usually a vasculitis triggered by acute SARS-CoV-2 infection. In this phase, the patient responds better to corticosteroids and the intubation time is usually shorter since there is an evident autoimmune component. Differentiating between acute COVID-19 or COVID-19 in a critical phase (cHIS) is essential, as the pathophysiology of the disease changes: Acute COVID-19: viral action. Chronic COVID-19: Immunosuppression and autoimmunity phase. Two data are necessary to understand whether the case is acute or not: 1- To ask the patient whether he has had respiratory decompensations in the last months and what the recurrence is or whether there has been any change in his behaviour or pain, or tiredness in the last months - this can mean COVID-19 that has not been previously diagnosed; 2- Observing tomography: heavy hard images with laminar atelectasis and thickening of bronchi and bronchioles: Chronic phase; A lighter image of infiltration in frosted glass: acute COVID-19. The pattern of pneumonia after COVID-19 has been acute fibrinous and organizing pneumonia (AFOP) that is a histological pattern characterized predominantly by the presence of intra-alveolar fibrin in the form of fibrin "balls" within the alveolar spaces, with a patchy distribution, and organizing pneumonia. E-H: Patient, 46-years-old, HIV+ with viral load always undetectable and CD4+ always higher than 600, started skin lesions associated with pruritus without improvement. He mentions having seen three other doctors who considered Kaposi's Sarcoma hypothesis. However, when asked about his last semester, the patient reported an essential change in mood. I note that the lesions can be due to mastocytosis after COVID-19 or even vasculitis. We started Dexamethasone equivalent to a 250 mg dose of Methylprednisolone for 3 days and de-escalating the dose for another 5 days. It evolves on the first day with many tremors that improved after the second day. It also evolves with the improvement of skin lesions. I associated acetylsalicylic acid thinking about platelet activation in case of vasculitis and request new tests. I: Tomography shows ectasia of the ascending aorta, accentuation of the centrilobular stipple and presence of bronchial thickening; CD8 + = 270 and CD4 + = 197. Tomography findings appear in vasculitis and other diseases of the parenchyma. Possible diagnoses: Vasculitis, mastocytosis. The patient will be reassessed for new Cd4+ and CD8+ and skin biopsy collections if there are new lesions. J-L: Young, male, 19-years-old. Admitted to HSPE due to dyspnoea for more than 8 days. IgM and IgG positive. At the time, we questioned granulomatosis with polyangiitis, but we were surprised to see a positive sputum smear for Mycobacteria tuberculosis. Reanalysing, this case should be recalled and seen by a new aspect: COVID-19 + induced vasculitis + activation of latent tuberculosis by immunoparalysis.
View Figure 6
Figure 6A-D: After ICU stay and orotracheal intubation and readmitted in the Infectious Diseases wards. Obese and or Diabetic patients after 14 days of disease. (14-40 days). Observe the "heavy" pattern of the images with opacities denoting extensive posterior atelectasis or even condensation in the pulmonary parenchyma in a diffuse manner. This tomography is different from the acute COVID-19 in Figure 1, where we have a ground grass pattern that is lighter and more tenuous. We are readmitting patients who already arrive with a "heavy" image pattern with new decompensation. This patient is not acute COVID-19; he is only exacerbating a disease, usually a vasculitis triggered by acute SARS-CoV-2 infection. In this phase, the patient responds better to corticosteroids and the intubation time is usually shorter since there is an evident autoimmune component. Differentiating between acute COVID-19 or COVID-19 in a critical phase (cHIS) is essential, as the pathophysiology of the disease changes: Acute COVID-19: viral action. Chronic COVID-19: Immunosuppression and autoimmunity phase. Two data are necessary to understand whether the case is acute or not: 1- To ask the patient whether he has had respiratory decompensations in the last months and what the recurrence is or whether there has been any change in his behaviour or pain, or tiredness in the last months - this can mean COVID-19 that has not been previously diagnosed; 2- Observing tomography: heavy hard images with laminar atelectasis and thickening of bronchi and bronchioles: Chronic phase; A lighter image of infiltration in frosted glass: acute COVID-19. The pattern of pneumonia after COVID-19 has been acute fibrinous and organizing pneumonia (AFOP) that is a histological pattern characterized predominantly by the presence of intra-alveolar fibrin in the form of fibrin "balls" within the alveolar spaces, with a patchy distribution, and organizing pneumonia. E-H: Patient, 46-years-old, HIV+ with viral load always undetectable and CD4+ always higher than 600, started skin lesions associated with pruritus without improvement. He mentions having seen three other doctors who considered Kaposi's Sarcoma hypothesis. However, when asked about his last semester, the patient reported an essential change in mood. I note that the lesions can be due to mastocytosis after COVID-19 or even vasculitis. We started Dexamethasone equivalent to a 250 mg dose of Methylprednisolone for 3 days and de-escalating the dose for another 5 days. It evolves on the first day with many tremors that improved after the second day. It also evolves with the improvement of skin lesions. I associated acetylsalicylic acid thinking about platelet activation in case of vasculitis and request new tests. I: Tomography shows ectasia of the ascending aorta, accentuation of the centrilobular stipple and presence of bronchial thickening; CD8 + = 270 and CD4 + = 197. Tomography findings appear in vasculitis and other diseases of the parenchyma. Possible diagnoses: Vasculitis, mastocytosis. The patient will be reassessed for new Cd4+ and CD8+ and skin biopsy collections if there are new lesions. J-L: Young, male, 19-years-old. Admitted to HSPE due to dyspnoea for more than 8 days. IgM and IgG positive. At the time, we questioned granulomatosis with polyangiitis, but we were surprised to see a positive sputum smear for Mycobacteria tuberculosis. Reanalysing, this case should be recalled and seen by a new aspect: COVID-19 + induced vasculitis + activation of latent tuberculosis by immunoparalysis.
View Figure 6
Lung biopsies and anatomopathological studies have shown an organising pneumonia pattern, with polymorphonuclear cells and fibrinous pneumonia with vessel necrosis, with more frequent or fewer eosinophils spectra of EGPA and GPA, as can be observed in Figures 4a-c and Figures 6j-l, and an Appendix showing different neutrophil orbit. These findings provide evidence that neutrophils have a different activation way from eosinophils, but both their orbits may sporadically collide. We can infer that there can be a substantial clinical variation to a condition like eosinophilic myocarditis leading to a heart failure clinical presentation, whose magnetic resonance image pattern, mimics that of viral myocarditis. The real diagnosis will be spoiled only by a biopsy - or due to a high level of suspicion of systemic eosinophilic disease. We have been following these patients after hospital discharge due to Covid-19. Their main complaint, in the vast majority of cases, is muscular pain and fatigue. Myalgia and eosinophilia syndrome with chronic fatigue is related to the tryptophan metabolite cascade alterations. The magnitude of the syndrome may be hypoxemia dependent. The longer time under hypoxemia, the less niacin and the more tryptophan will deviate to Kynurenine in an inflamed environment (Figure 7 - Appendix).
 Figure 7a: Credits
Figure 7a: Credits
Herr, N., Bode, C., & Duerschmied, D. (2017). The Effects of Serotonin in Immune Cells. Frontiers in cardiovascular medicine, 4, 48. https://doi.org/10.3389/fcvm.2017.00048
View Figure 7a
 Credits
Credits
Nora Torok, Rita Torok, Zoltan Szolnoki, Ferenc Somogyvari, Peter Klivenyi, Laszlo Vecsei, "The Genetic Link between Parkinson's Disease and the Kynurenine Pathway Is Still Missing", Parkinson's Disease, vol. 2015, Article ID 474135, 7 pages, 2015. https://doi.org/10.1155/2015/474135
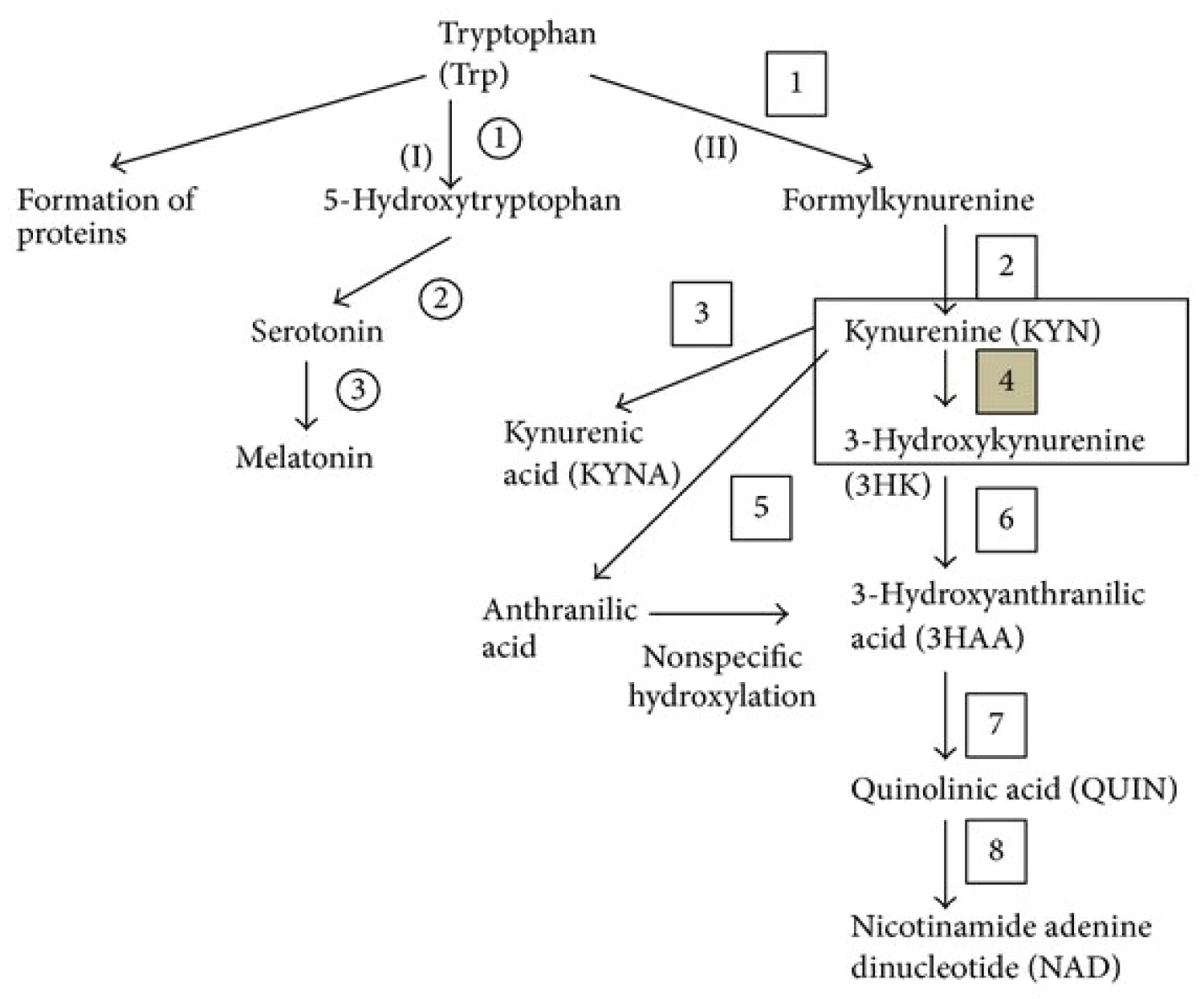
Figure 7B: Tryptophan metabolism. Kynurenine monooxygenase (KMO) (number 4) is an enzyme of the kynurenine (Kyn) pathway (KP), which is the major catabolic route of tryptophan. Kyn represents a branch point of the KP, being converted into the neurotoxin 3-hydroxykynurenine via KMO, neuroprotectant kynurenic acid, and anthranilic acid. As a result of this branch point, KMO is an attractive drug target for several neurodegenerative and/or neuroinflammatory diseases, especially Huntington's (HD), Alzheimer's (AD), and Parkinson's (PD) diseases.
(I) Serotonin pathway: 1: Tryptophan hydroxylase, 2: L-aromatic amino acid decarboxylase, and 3: Serotonin-N-acetyltransferase and hydroxyindole-O-methyltransferase; (II) kynurenine pathway: 1: Tryptophan dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO), 2: Formamidase, 3: Kynurenine aminotransferases (KATs), 4: Kynurenine 3-monooxygenase (KMO), 5: Kynureninase, 6: Kynureninase, 7: 3-hydroxyanthranilic acid oxygenase, and 8: Quinolinic acid phosphoribosyltransferase.
View Figure 7b
Treatment using methylprednisolone pulse injections in the perfect timing has blocked the evolution towards intubation in the acute phase, by preventing eosinophilia. Problem is: When is the perfect timing? Empirically and based on publication reviews, we have defined the best period for this intervention between the seventh and eighth days after the beginning of symptoms, and while the patient still shows arterial oxygen relation higher than 200 (PaO2/IFO2 > 200) with chest tomography studies showing bilateral lung parenchymal injury. Preferably, we indicate this pulse therapy before C-reactive protein dosing reaches 10 mg/L.
At this moment, the HLA-DR expression is either still at its baseline levels or still a little repressed and the IL-6 secretion is elevated - but still within a functional range. Pulse therapy in this phase is crucial for patients not to progress and need intubation performing. However, even if the pulse therapy was performed and the patient's clinical presentation or blood gas analysis worsen, intubation should not be delayed or postponed.
The latest phase of the disease is represented by multiple spectral diseases. These patients should be treated following adequate conditions, like organising and fibrinous pneumonia, and eosinophilic vasculitis. Corticosteroid-based long-term treatments should be considered, and other immunosuppressive drugs may be associated (Appendix) or in low doses. However, we have been using pulse doses according to Tehran's protocol. Nowadays, we have some doubts about corticosteroids indication when considering HLA-DR expression (the low expression is likely to have less response to corticosteroids), and are pronated to consider alternative immunosuppressive therapies, immunoglobulin administration or plasmapheresis. But never to diabetics and obese.
There are still more unanswered problems we need to deal. Neoplastic transformation is very plausible: Eosinophilia, mastocytosis neutrophil overstimulation, angiogenesis, and tolerance angiogenesis. This last may allow the growth of tumours that were "in silence", in an environment with NK cells responsible for killing tumoral cells. An assessment must be done to show if COVID19 has been allowing rapid development of neoplasms sometimes coincidences become statistics when there is a pattern of repetition. Therefore, we observed the frequent neurological symptoms: It is unusual to find so often behaviour changes or consciousness lowering. The cause is either reactivated infections or CNS inflammation or even CNS eosinophilic reaction. Today, our only option is performing a complete screening, with at least mandatory chest CT scans for any patient presenting any consciousness alteration, myocardial infarction, stroke, or thrombosis. All these hypotheses based on our clinical experience and correlation with laboratory findings (data which we have been organising, but we have been developing high intense Laboral activities that work looking at these data have been being almost impossible, even so, we have this important relationship to incite a balance on the closed eyes of professionals that need to feel that COVID-19 is not a single disease.
Immunoparalysis explains why there is so much variance in IgG production. Variety of symptoms and the immunological variances in patients in the same group are much different and without a pattern. B cells produce IgM independently of TCD4+ cells, but to produce IgG, B cells necessarily need TCD4+ function and have an epitope presented by antigen-presenting cells. As patients have different degrees of immunosuppression, we have different forms of IgG kinetics.
Another marking phenomenon is the observed cyclic nature of neutrophilia. Every five to seven days, the patient's blood counts will show this increase. Coincidentally, platelets' half-lives are 5 to 7 days. In every platelet activation period or every cell death period, there is a marked Ca2+ requirement. All the observed patients showed a serum calcium lowering pattern every 5 to 7 days - sometimes, with such intense variations that could be found through electrocardiographic changes and sudden death risk. Calcium plays a fundamental role in neuromuscular physiologic mechanisms, and its lowering (hypocalcaemia) is known both to impair myocardial contractility and to prolong the QT interval, predisposing to ventricular arrhythmias.
The most common explanation for hypocalcaemia in critically ill patients is hypoalbuminemia - and this usually does not require calcium correction, but nutritional optimisation. Hypocalcaemia signs and symptoms include mental status alterations, tetany, positive Chvostek and Trousseaus signs, cardiac arrhythmias, laryngospasm, and eventually hypotension. Hypocalcaemia seems to be explained by tissue chemotaxis via an inflammatory pole triggered by autoantibodies, IL-6, Kynurenine neutrophil chemotaxis and the complement system, as in EGPA and GPA for 5-7 days increasing cells up to a suddenly falls [59,76-78,82]. In this interval, it is possible to observe a daily increase in the leukocytes counting in the blood c, with slow and progressive young forms deviation (Figure 4b). When the patient reaches a new peak of generalised discomfort, his laboratory studies begin to show an increase in transaminases, lipase, amylase, and he experiences respiratory worsening, which is proof of new and repeated inflammation. Simultaneously, there should be an increase in interferon-gamma (INF-γ) to decrease neutrophilia. INF-γ further stimulates monocytes to shift the axis to IL-6 at this stage of the disease, and the cycle is indefinitely perpetuated. Each cycle is associated with more injury, more angiogenesis, more factors that trigger autoimmunity, more thrombosis and hypocalcaemia due to consumption.
Moreover, there is something even worse. People in the asymptomatic infection are also included in this syndrome scenario - even in more discrete and without many symptoms, but yet there they are. Some followed outpatients who never had symptoms of COVID-19 have been reported to show odd symptoms, like diarrhoea, bone pain or neuropathic pain. Figure 6e-h refers to an outpatient presenting mastocytosis and mood disorders - the only altered parameter in basic exams is a monocytosis with leukopenia found in his blood cell count, with normal eosinophil counting and mild laminar atelectasis and bronchial thickening visible at chest CT scan. The lesson learned: Always investigate.
COVID-19 has an intense ability to trigger an autoimmune (or neutrophilia induced by tryptophan - Appendix) response started by NET formation; thus, patients who were asymptomatically infected or even who do not think they had a disease deserve a thorough clinical evaluation, even with negative serologic studies. SARS-CoV-2 may have gone unnoticed, and only later will symptoms appear - when no one can explain. Be aware of mastocytosis-like skin lesions, as they are reasonable indications of some subclinical immune process that may be happening and repetitive pictures of airway infection.
The spectra of chronic COVID-19 are broad and hidden. We are going through a highly contagious pandemic, and it is barely impossible not to get infected.
Another observed vital point to be raised is that possibly any medication acting on neutrophils can minor the intensity of Covid-19 symptoms. Some clozapine using patients, as well as other psychiatric drugs that usually causes some neutropenia had mild symptoms that did not evolve to severe forms of the disease. Baseline neutropenic presenting patients - either due to hypothyroidism, prolonged use of insulin or other medications in use, usually showed only mild disease manifestations. From this point of view, cirrhotic patients are a population at risk of being underdiagnosed for COVID-19. These patients have basal deficient and lagging neutrophils and liver factors that attract these cells, usually have been found manifesting milder forms of the disease and end up receiving diagnoses of hepatic encephalopathy in case of lowering of consciousness. However, hypoxemia and viral cause can be underlying. I would like to include in this same theory the idea that patients receiving HIV antiretroviral drugs also show minor symptoms, and many antiretrovirals cause some bone marrow toxicity, compromising the activation of neutrophils (Appendix).
This article was the framework formed from many readings and daily observations of COVID-19 patients. A daily struggle and an unforgettable learning that I dedicate to all those who unfortunately died due to this terrible and unknown disease.
1. Are cases of prostate cancer increasing because the prostate has a high number of ACE-2 receptors?
2. May Cases of reactional leucocytosis of the inflammatory syndrome be confused with leukaemia?
3. Can post-COVID-19 leukopenia be confused with bone marrow aplasia?
4. There is a massive denial of listing COVID-19 as a possible diagnose among the Hypotheses possibilities when a case is discussed. Would it be explained by fear and denying? Is it the existence of a single virus that causes so many disorders inside and outside the human body that remind them that they are fragile like other species? Is to deny COVID-19 the solution? Alternatively, Is it the solution to our problems in the face of the fear of recognizing that we know nothing and we are nothing and that Sars-CoV-2 imposed on human beings the new time of disease evolution, social and behavioral changes. We have been living in captivity for the virus for a year.
5. Laboratory tests for IgM and IgG detection are said to be low quality. What happens is low production of antibodies due to the triad's scarcity to produce IgG: B cells, TCD4+ cells and antigen-presenting cell (APC). Based on this scenario, what is the reason for not performing IgA Search as a screening method?
6. Many people stay for a long time in immunoparalysis, a variable time syndrome for each person. Besides, the hypothesis that inflammatory syndrome is related to autoimmune diseases is increasingly gaining ground. It is essential to ask: Do vaccines injected in an immunoparalysis phase provide antibody formation against the epitopes contained in its formulation?
7. Many SARS-CoV-2 previously infected may have produced a low antibody titer to the point of not being identified in laboratory tests. These were considered "virgins" of infection, while they may have antibodies at low titers and decreasing memory cells. Considering this reality:
8. Was the first dose, first of all, of the vaccine a booster in these pre-exposed people?
9. Vaccines could trigger or increase autoantibodies' formation, as it was injected in people who have already been exposed to the virus? Does Tryptophan syndrome cause everything after acute COVID-19?
10. Is everything caused by Tryptophan syndrome after acute COVID-19?
To Augusto Yamaguti - Head of HSPE Infectious Diseases, Andrea Lucia Ladeira de Almeida - Clinical manager of HSPE and Ederlon Alves de Carvalho Rezende - Head of ICU HSPE, thank you for believing in me when I needed people to believe in me.
To the HSPE Infectious Diseases physicians: Adriana Macedo Dell 'Aquila, Antonio Mitihossi Nagamachi, Augusto Yamaguti, Caio Rosenthal, Carlos Armando de Ávila, Cibele Lefevre Correa Fonseca, Cinzia Trevisanello, Cristiano Melo Gamba, Daniela Kallíope de Sá Paraskevopoulos, Davi Francisco Lopez, Durval Alex Gomes e Costa, João Silva de Mendonça, Marcelo Milleto Mostardeiro, Marina Keiko Kwabara Tsukumo, Marli Sasaki, Rosa Maria Barbosa,Thaís Guimarães, Vera Maria Coutinho de Moraes, Zilda Zuleima e Silva, Residents in Infectious Diseases: Amanda Fernandes Silva Takenaka, Ana Flavia Forato Pereira, Letícia Verona Martinis Costa, Marcela Gonsales Menis, Beatriz Turato Mendonça, Luisa Caracik de Camargo Andrade, Gabrielle Picanco Rilhas. Thank you for everything and especially for constant learning because even with different thoughts (which is good), I always felt comforted by you.
Technical director of HSPE: Dr. Kátia Antunes
Clinical director: Lilia Azzi Collet da Rocha Camargo
Research Centre 2 - Lilian Ferrari, Natália Cerqueira, Deni Gomes, Carol Cardona, Ney, and Zelinda Bartolomei and the whole team with whom I always learn. LIM-60 FM - USP: Priscilla, Cassia, Carol A, Carol Soares, Matheus, Juliana, Mariana, Amanda. My friends that every moment understand my immune brain storming. I dedicate this To family and friends for tolerating my absence in the COVID-19 period and giving me the strength to always move on and to my teacher Esper Kallás: For your ethics as a person and your ethics in research and in Medicine.
Debora Sitnik. Her at a glance on this work, she organized my ideas because she has the power in understanding how my head works. My teacher, my friend.
*"COVER-ME-UP": Pulsoterapia com Metilprednisolona em pacientes COVID-19: Estudo randomizado e controlado. COVID-19: A randomized study using methylprednisolone in pulse therapy.
Manuscript has approved by HSPE Research Ethics Committee.
According to the National Research Ethics Commission of the Federative Republic of Brazil - CNSResolution No. 466 of 2012, this review does not require a consent form, as it does not present data that directly identify patients and because it is a theoretical research that it arises naturally from a professional practice that uses data that does not reveal the subjects/patients. Even so, the article is filed with the HSPE RESEARCH ETHICS COMMITTEE, via the Brazil research platform with approved dismissal.
Manuscript has approved by HSPE Research Ethics Committee.
No funding was received.
All data are available in the electronic medical record system of Hospital do Servidor Estadual (Sao Paulo - Brazil) and in the authors' research files.
Luiz Zanella wrote this article and elaborated all the Figures based on the Biorender platform. Performing an active participation in the management of COVID-19 patients.
Luciana Galvão is a key person that supports many of the ideas contained in this article. She elaborated many hypotheses, built concepts and has an active role in the management of COVID-19 patients. We are principal investigators "COVER-ME-UP" TRIAL (Methylprednisolone pulse).
None of authors have competing interests.
This article aims to expose:
- That COVID-19 is a chronic disease.
- The acute phase of the disease is viral and inflammatory, with the inflammatory component being responsible for severe acute respiratory failure.
- The chronic disease is due to changes in the metabolism of tryptophan and the lack of niacin (NAD/NADH+). Tryptophan has its metabolism altered by the lack of intestinal absorption due to internalization of ACE-2 and hypoxemia and inflammation, diverting its products to the formation of toxic Kynurenine metabolites.
- cHIS is pan-syndromic, the most evident being IL-6-mediated inflammatory syndrome, immunoparalysis syndrome, variable hypereosinophilic syndrome.
- The chronic phase (cHIS) can present multiple systemic changes such as muscle pain, skin lesions, changes in the central nervous system due to the presence of mastocytosis and eosinophilia, and the toxic component of tryptophan products.
- The formation of antibodies does not follow a pattern of formation since the disease is specific to each person and dependent on the immunosuppression that the virus determined for each individual.
- In the immunoparalysis phase, we can reactivate latent infectious diseases such as fungal and bacterial infections such as mycobacteriosis.
- The pneumonia pattern in the chronic phase is an inflammatory component and is a disease already described called acute fibrinous organization pneumonia (AFOP) secondary to IL-6 and sustained inflammation.
- COVID-19 is a viral disease and, like other viral infections, can also cause diseases of autoimmune origin. Perhaps its propensity for autoimmunity to occur is that the virus triggers autoantibodies' formation while endothelial damage occurs.
- That COVID-19 causes a tolerance profile facilitating the appearance of autoimmune diseases and neoplasms.
- That the inflammatory component is dependent, among other factors, on the length of hypoxemia to which a person has been subjected in the acute phase of the disease with consequent consumption of NAD/NADH+ and stimulation of neovasogenesis.
- That there is a concomitance of immunological forces of Treg and Th17 profile.
- Persistent anaemia is due to the continuous activation of neutrophils in the subacute and chronic phases and dependent on the serotonin/tryptophan pathways.
- That COVID-19 in the cHIS phase presents an oscillating (almost sinusoidal) pattern of laboratory behaviour indicating periods with an increase followed by a sudden decrease in cell types/proteins always marked by hypocalcaemia.
- That Ca2+ is necessary for activation of neutrophils, cellular apoptosis and many other cellular metabolic systems is very consumed.
- Bone metabolism is greatly affected by serotonin and mastocytosis, which is why fractures and osteoporosis may increase after COVID-19.
- That both the first and second phases respond to corticosteroids.
- That COVID-19 still has a lot to teach us.