Pleomorphic xanthoastrocytoma (PXA) is a rare tumor with favorable prognosis, classified as grade II in the World Health Organization (WHO) and accounts for less than 1% of all astrocytic neoplasm. It is commonly found in childhood and young adults. This tumor has been described as part of the spectrum of Long-Term Epilepsy Associated Tumors (LEAT). The most common location is supratentorial, involving predominantly the temporal lobe. Here we present the case of a 6-year-old female with long-term, drug-resistant, complex epilepsy of the frontal lobe, associated with a frontal PXA in which total remission of seizures was achieved after surgical resection. The objective of this paper is to highlight the importance of PXA in the differential diagnosis of long term epilepsy in children.
Pleomorphic xanthoastrocytoma, Anaplastic, Glioma, Drug-resistant epilepsy
Pleomorphic xanthoastrocytoma (PXA) is a rare tumor with favorable prognosis, classified as grade II in the World Health Organization (WHO) and accounts for less than 1% of all astrocytic neoplasms [1,2]. It is commonly found in childhood and young adults [3]. There is no reported prevalence in the Mexican population because, as in the world, few cases exist in the literature. Gonzálo-Solis, et al. reported a case about a Mexican patient who debuted with hemianopsia and hypertensive encephalopathy [4]. PXA is usually located in the supratentorial compartment, involving predominantly the temporal lobe (43%), followed by frontal and parietal lobes [3,5]. The etiology of PXA is unknown. Rarely, PXAs have been reported in patients with neurofibromatosis type 1 [6], but the majority arise sporadically without any evidence of genetic predisposition [7]. Seizures are the most common symptom of PXA. Furthermore, this tumor has been described as part of the spectrum of Long-Term Epilepsy Associated Tumors (LEAT), which involves lesions identified in patients with two or more years of drug-resistant epilepsy [8].
A 6-year-old female with a two-year history of idiopathic complex focal epilepsy treated with Levetiracetam 1 gr/day and Magnesium Valproate 1.2 gr/day was admitted to emergency department for presenting seizures lasting 15 seconds, every 2 hours, consisting of laugh/cry episodes accompanied by rhythmic pelvic movements and a tonic position of both upper limbs (beginning with right upper limb) lasting 3 seconds, followed by clonic movements. During emergency department stay, seizures became continuous despite anticonvulsant IV therapy; sedation was initiated, and an electroencephalogram (EEG) reported non convulsive status epilepticus.
Brain CT scan revealed a poorly demarcated hypodense zone in the left frontal lobe (Figure 1). MRI showed a 1.6 × 1.5 × 1.5 cm, homogeneously enhanced lesion within the subcortical region of the left superior frontal gyrus with compression of the cingulate gyrus and supplementary motor cortex (Figure 2a and Figure 2b). The spectroscopy shoed an altered choline/creatinine ratio and decreased NAA, suggestive of a glial tumor (Figure 2c).
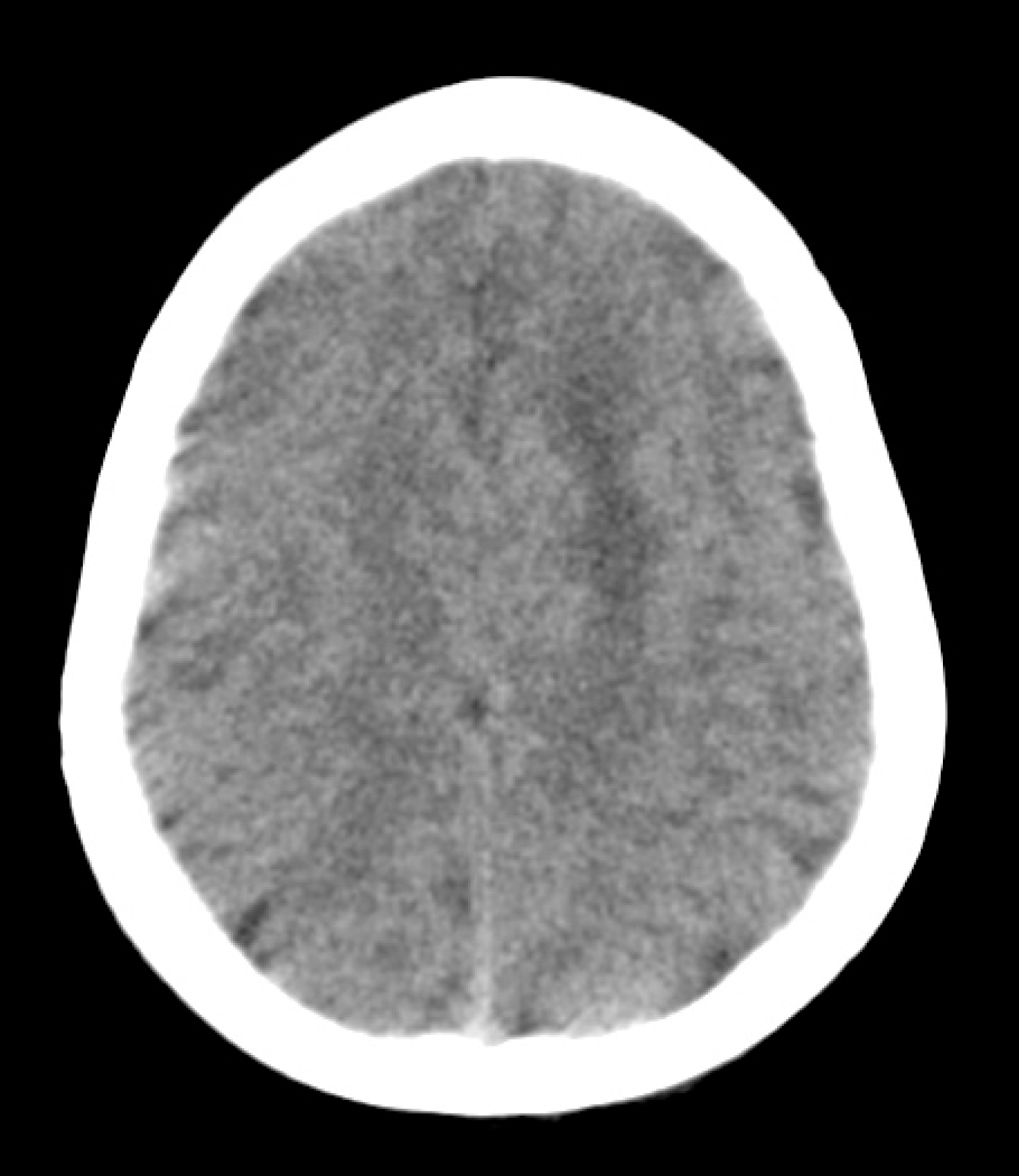 Figure 1: Non-contrasted brain CT scan. Axial cut showing a poorly demarcated hypodense zone in the left frontal lobe.
Figure 1: Non-contrasted brain CT scan. Axial cut showing a poorly demarcated hypodense zone in the left frontal lobe.
CT = Computed Tomography.
View Figure 1
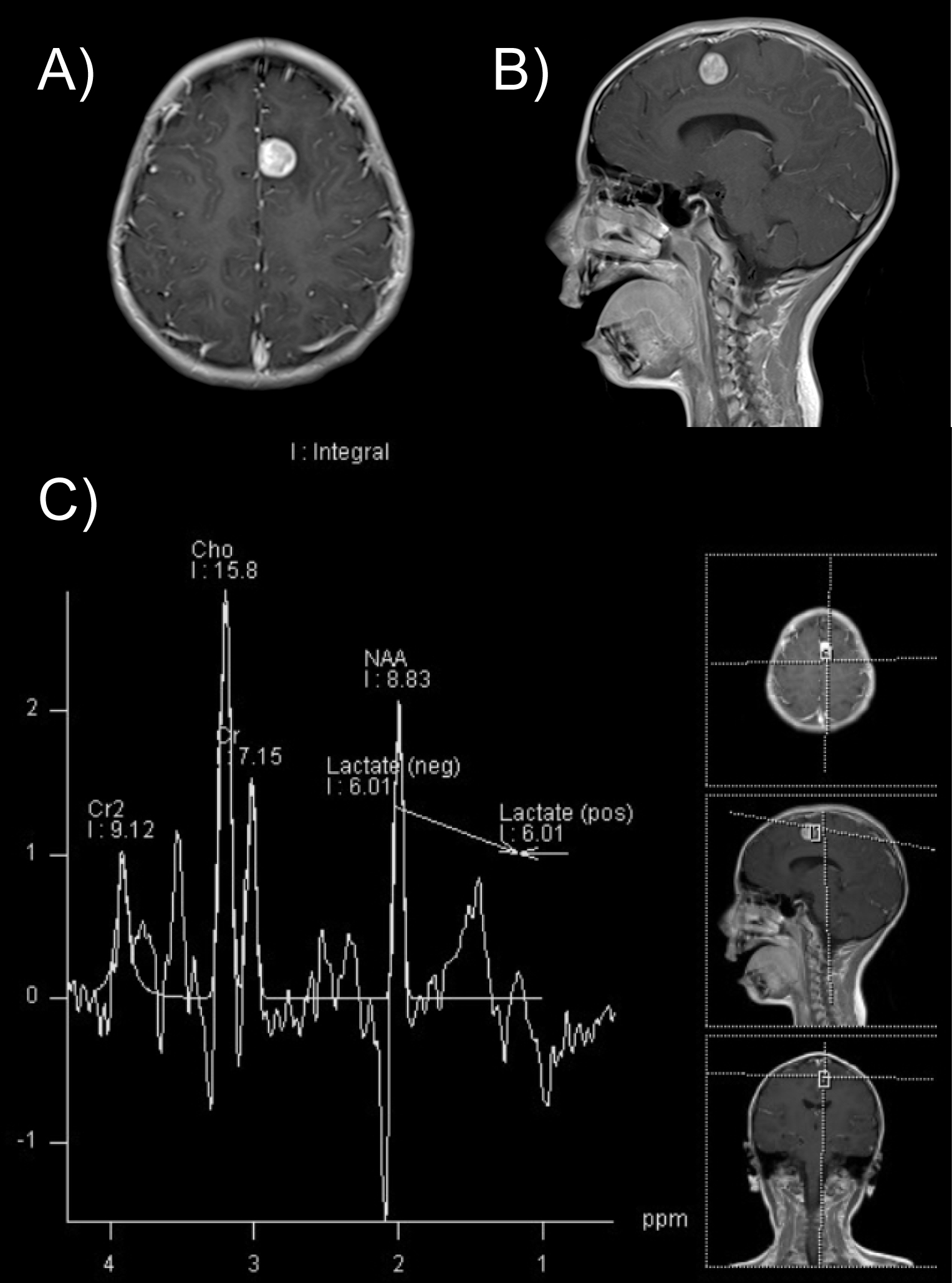 Figure 2: T1-weighted gadolinium enhanced MRI (A) Axial cut showing a homogeneously enhanced lesion in the subcortical region of the left superior frontal gyrus; (B) With compression of the cingulate gyrus and supplementary motor cortex; (C) Spectroscopy. Abnormal choline/creatinine ratio and decreased NAA. Highly suggestive of a glial tumor.
Figure 2: T1-weighted gadolinium enhanced MRI (A) Axial cut showing a homogeneously enhanced lesion in the subcortical region of the left superior frontal gyrus; (B) With compression of the cingulate gyrus and supplementary motor cortex; (C) Spectroscopy. Abnormal choline/creatinine ratio and decreased NAA. Highly suggestive of a glial tumor.
MRI: Magnetic resonance imaging; NAA = N-acetyl aspartate.
View Figure 2
A directed craniotomy was performed and a 100% resection of the lesion was achieved without complications. The immediate postsurgical course was satisfactory, without neurological deficit or seizures. The post-operative MRI (Figure 3) showed expected postsurgical changes and no evidence of residual tumor.
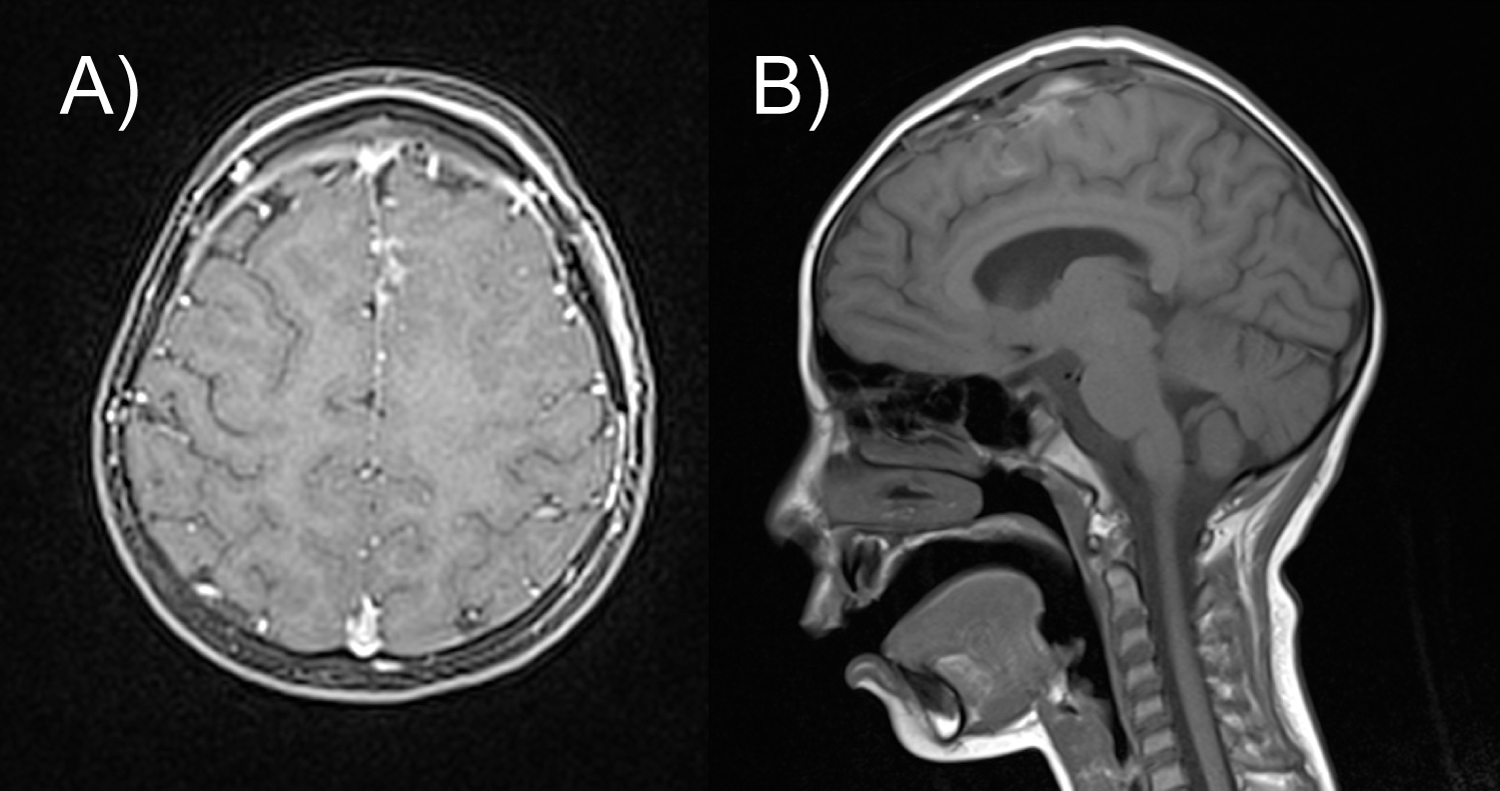 Figure 3: Postoperative T1-weighted MRI (A) Axial cut; (B) Sagittal cut showing complete resection of the tumor and a residual hyperintense image in relation to minor postoperative bleeding and a surrounding hypointense image corresponding to encephalomalacia.
View Figure 3
Figure 3: Postoperative T1-weighted MRI (A) Axial cut; (B) Sagittal cut showing complete resection of the tumor and a residual hyperintense image in relation to minor postoperative bleeding and a surrounding hypointense image corresponding to encephalomalacia.
View Figure 3
Immunohistological analysis of the lesion (Figure 4) revealed a tumor compatible with a WHO Grade II Pleomorphic Xanthoastrocytoma; positive to glial fibrillary acidic protein (GFAP), synaptophysin and CD43; negative to epithelial membrane antigen.
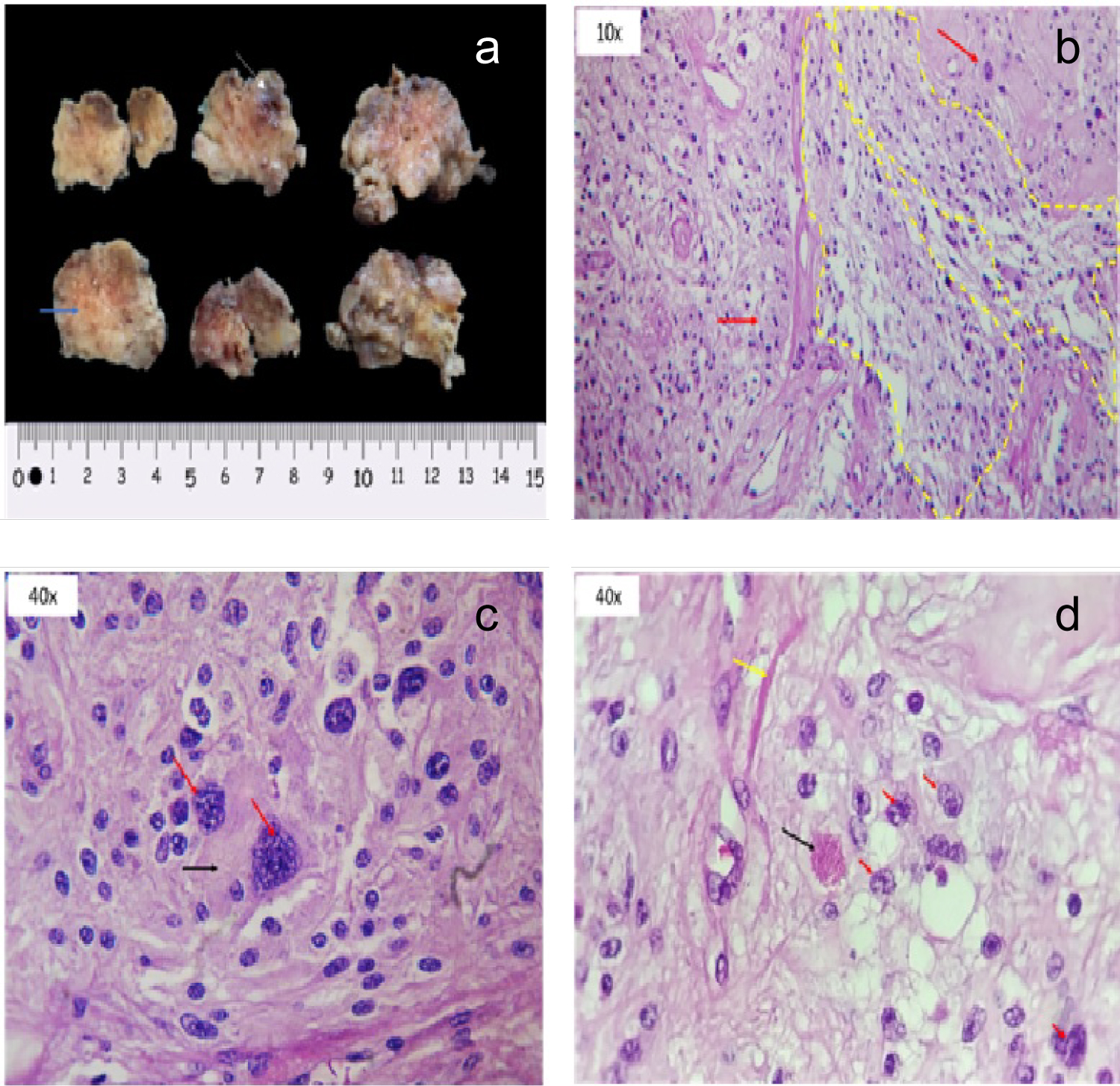 Figure 4: Histological analysis (a) Macroscopic view of the specimen. Irregular surface with meaty appearance (blue arrow) which alternates with hemorrhagic zones (white arrow); (b) 10x microscopy demonstrating a disorganized architectural pattern. Some neoplastic cells show a fasciculate disposition (shown inside the yellow lines). Red arrow show net-organized cells. Both patterns over a fibrillar base characteristic of astrocytic neoplasms; (c) 40x microscopy pointing with red arrows two pleomorphic astrocytic cells. Abundant eosinophilic cytoplasm (black arrow) simulates xanthocyte cytoplasm; (d) 40x microscopy showing secondary neoplastic characteristics. Irregularities of nuclear edge (red arrows), eosinophilic granules (black arrow) and Rosenthal fibers (yellow arrow) are seen.
View Figure 4
Figure 4: Histological analysis (a) Macroscopic view of the specimen. Irregular surface with meaty appearance (blue arrow) which alternates with hemorrhagic zones (white arrow); (b) 10x microscopy demonstrating a disorganized architectural pattern. Some neoplastic cells show a fasciculate disposition (shown inside the yellow lines). Red arrow show net-organized cells. Both patterns over a fibrillar base characteristic of astrocytic neoplasms; (c) 40x microscopy pointing with red arrows two pleomorphic astrocytic cells. Abundant eosinophilic cytoplasm (black arrow) simulates xanthocyte cytoplasm; (d) 40x microscopy showing secondary neoplastic characteristics. Irregularities of nuclear edge (red arrows), eosinophilic granules (black arrow) and Rosenthal fibers (yellow arrow) are seen.
View Figure 4
At 1-year follow-up, the patient is in management with Levetiracetam 1 gr/day and Magnesium Valproate 400 mg/day and remains seizure-free, with a normal neurological exam and an adequate neuropsychological growth. No adjuvant therapy was needed. Tumor recurrence has been monitored with serial MRIs (Figure 5), with no evidence of relapse. No follow-up EEG has been made.
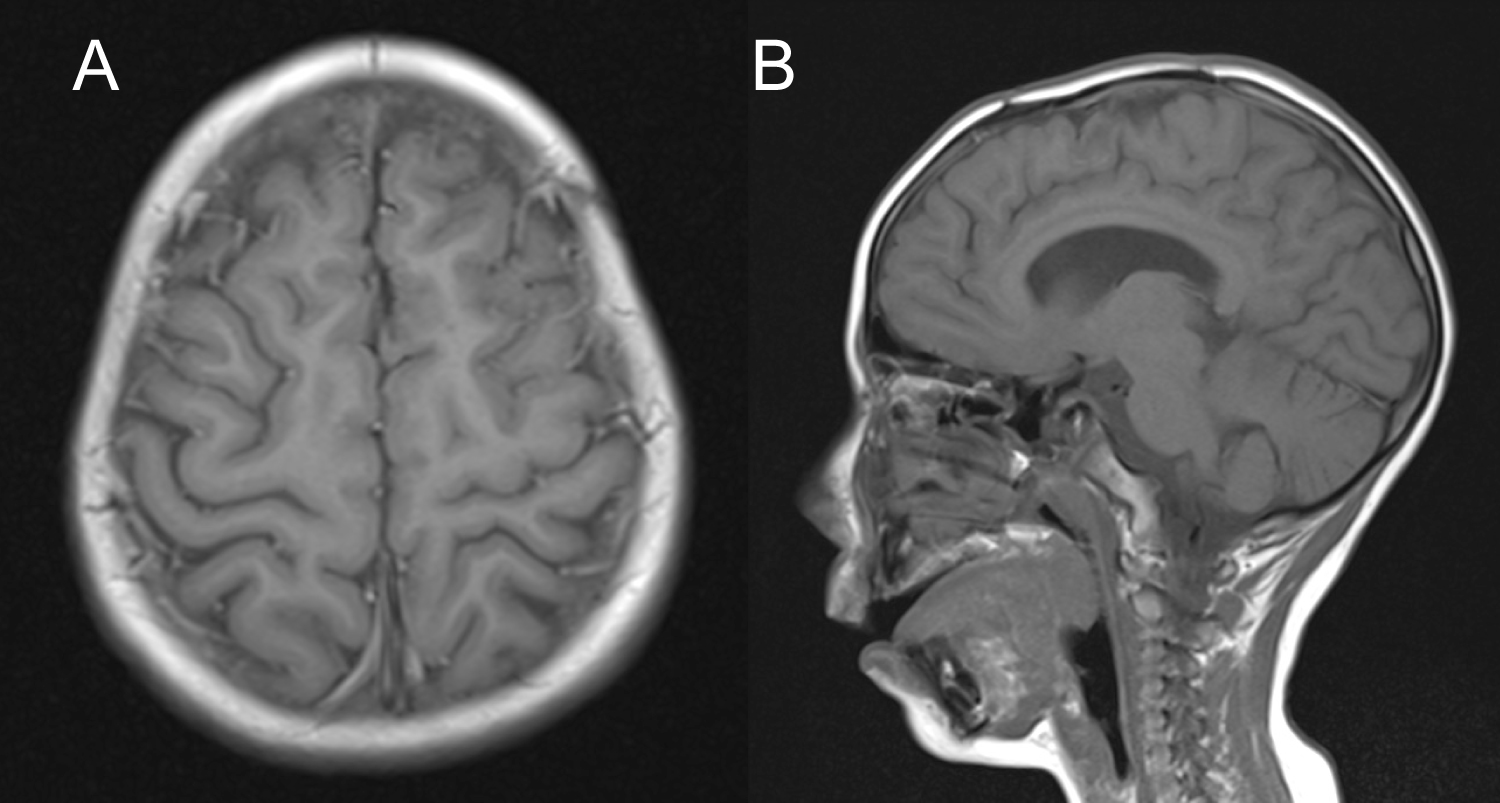 Figure 5: Follow-upT1-weighted MRI (A) Axial cut; (B) Sagittal cut showing no evidence of recurrent tumor and a minor hypointense image in the surgical site corresponding to encephalomalacia.
View Figure 5
Figure 5: Follow-upT1-weighted MRI (A) Axial cut; (B) Sagittal cut showing no evidence of recurrent tumor and a minor hypointense image in the surgical site corresponding to encephalomalacia.
View Figure 5
PXA is an uncommon tumor that accounts for less than 1% of all astrocytic neoplasms [1,2], commonly found in childhood and young adults with mean age presentation of 22 years [3]. The most common symptom related to this tumor are seizures (71%), which have been described as long-lasting (median of 3 years), reported as being part of the LEAT spectrum [3,8]. The most common location is supratentorial (97%), involving the temporal lobe in 43% (35% exclusively temporal lobe and 8% with > 1 lobe including temporal lobe) [5]. PXAs located in the frontal lobe accounts for approximately 10% of all cases [3] (Table1).
Table1: Location frequency in PXA [3,5]. View Table 1
In a retrospective study of 25 patients in which presenting symptoms were known in 17 patients, seizures were the most common manifestation. However, seizures were present in less than half (47%) of the patients. Another 41% of patient symptoms were related to increased intracranial pressure. In addition, the main clinical manifestations in patients with frontal lobe PXA were: seizures, focal neurological deficit, nausea, vomiting, and headaches [9].
In another study of 13 patients with PXA confirmed by histopathology, the main clinical manifestations were: Seizures (the most frequent, n = 8) and cephalalgia (n = 5), being the frontal location (n = 1) and frontoparietal (n = 1) of low frequency and associated with seizure symptoms [10].
As in the vast majority of glioneuronal tumors, long-term focal complex epilepsy that is often resistant to pharmacological therapy has been reported as the main type of epilepsy of PXA [11]. A case series of 15 patients with glioneuronal tumors reported that 66.6% of the patients had suffered complex partial seizures [12].
The patient was diagnosed 2 years earlier with idiopathic complex focal epilepsy, with partial control of the convulsive activity with Levetiracetam 1 gr/day and Magnesium Valproate 1.2 gr/day; parents reported two to three seizures per week with the characteristics already described before admission. CT scans of the brain performed as part of the diagnosis approach were considered normal (Figure 1) shows the CT scan performed before neurosurgical evaluation). Moreover, multiple EEGs, of which only its general characteristics could be documented, reported abnormal spike-and-slow-wave complexes in paroxysms of up to 10 seconds, without identification of a specific epileptogenic location.
Seizures originating near the supplementary motor cortex are characterized primarily by complex automatisms consisting of unmotivated laughter followed by tonic postures; seizures initiating in the medial frontal cortex and the cingulate gyrus consists of motor agitation, gesticulation and repetitive motor activity of the pelvis, usually associated with unmotivated laughter or cry [13].
NICE guidelines on epilepsies recommend EEG and brain MRI as part of the work-up of pediatric epilepsy, especially in focal and drug-resistant epilepsy [14]. MRI should be performed in all patients with epilepsy, as it is crucial for the diagnosis and treatment and allows characterization of the nature of different types of epileptogenic lesions. Neuroimaging is critical in the evaluation of newly diagnosed patients and, as mentioned above, in patients with refractory seizures, especially in patients with focal findings in the neurological physical examination. Given that MRI can help differentiate between progressive and static lesions, it is an important tool for prognostic counseling and treatment strategy [15].
EEG, despite being an important part of the evaluation of a patient with seizures, has its limitations when attempting to record activity from insular cortex, fronto-parietal opercular cortex, inferomedial temporal lobe, interhemispheric fissure, orbitofrontal cortex, inferior parietal-occipital cortices and deep sulcus [16]. This is consistent with the difficulty on identifying a specific epileptogenic location in our patient.
Brain MRI in PXA commonly illustrate a solid component isointense on T1-weighted images and hyperintense on T2-weighted images with marked enhancement after contrast administration [17,18]. In our case, brain MRI findings were similar to the typical imaging pattern described in the literature and were an important part of the work-up because of the underrepresentation of the brain activity on EEG given the tumor's location.
PXA is a WHO grade II tumor with 5-year survival rate of 90.4% and 5-year recurrence-free rate of 70.4% when treated. Thus, this tumor is considered to have favorable prognosis. The main treatment is surgery and the prognosis is directly proportional to the percentage of tumor resection [1]. One of the main concerns of patients with PXA is controlling the seizures; fortunately, PXA related seizures have been described as well-responsive to surgery. Different case series have reported Engel Class IA after total resection of PXA [11,12,19-21].
Histological features of PXAs include marked pleomorphic and lipidized tumor cells, eosinophilic protein droplets, prominent reactive lymphocytic infiltrates and a pericellular/perilobular network of reticulin fibers; tumor cells are immuno positive to GFAP, S100 protein, class III β tubulin and synaptophysin [22,23]. Recently, PXA has been found to harbor the highest frequencies of BRAF V600E mutation in primary central nervous system (CNS) neoplasms (up to 60%-78%). This mutation can be identified by immunohistochemistry using a BRAF V600E mutation-specific monoclonal antibody [24]. CD34 expression has also been described in PXAs and this finding may be useful in the differential diagnosis with diffusely infiltrating malignant gliomas [24]. Our patient's tumor immunohistological analysis revealed pleomorphic astrocytic cells with abundant eosinophilic cytoplasm (Figure 3b and Figure 3d) and was positive to GFAP, synaptophysin and CD34.
PXA should be suspected in patients with long-term drug-resistant epilepsy. The diagnosis of this tumor can be challenging given the brain CT scan minimal changes in the initial evaluation and especially when located in areas difficult to identify by EEG, as we report in this case. We point out the importance of MRI in epilepsy protocol and immunoanalysis to differentiate PXA from other more aggressive neoplasms, all as a comprehensive assessment to improve timely approach and grant a favorable prognosis to patients with PXA.
All authors have contributed equally to the work.
None to declare.
The authors declare no relevant funding or special scholarships from public, commercial, or non-profit bodies.