Objectives: To report a rare case of malignant transformation of a peripheral nerve sheath tumour (MPNST) with associated metastasis in a patient with Neurofibromatosis 2 (NF2).
NF2 is a hereditary disorder which presents with both central and peripheral nervous system tumours; caused by defects in the long arm of chromosome 22. The prevalence of NF2 is 1 in 50,000. NF2 is not a recognised risk factor for malignant transformation of schwannomas and this in this group is rarely seen, despite their high burden of schwannomas.
Case: A 30-year-old patient with known NF2 presented to John Radcliffe Hospital with right sided hip pain, anterior thigh pain and weakness in his legs that developed over 7 months, affecting his mobility. Multiple investigations including MRI scans and ultrasound guided biopsy revealed a malignant peripheral nerve sheath tumour with metastatic spread, arising from a longstanding L4 lesion, previously diagnosed radiologically as a schwannoma.
Conclusion: The case highlighted demonstrates a rare incidence of malignant transformation of a spinal schwannoma and outlines diagnostic and management approaches.
Malignant peripheral nerve sheath tumour, Neurofibromatosis type 2, Rare transformation
Neurofibromatosis 2 (NF2) is an autosomal dominant, hereditary disorder caused by defects in the long arm of chromosome 22 which lead to benign tumour growth [1]. Prevalence of NF2 is 1 in 50,000 [2], with patients developing both central and peripheral nervous system tumours [3]. Vestibular schwannomas are most commonly seen (95% of patients) and the presence of bilateral vestibular schwannomas is diagnostic. NF2 is also associated with meningiomas, gliomas, cerebral calcification, posterior subcapsular lens opacity and schwannomas in nerves other than cranial nerve VIII. 90% of patients will develop schwannomas in the spine [3]. Malignant transformation of schwannomas is rarely seen. If by some small chance malignant change does occur, it is usually to neurofibrosarcoma [4]. In this report, a rare case of malignant transformation of a schwannoma in a patient with NF2 is described.
A 30-year-old male patient was diagnosed with NF2 as a child, at the age of 9 years after his father was found to have NF2. His genetic severity score was 2A/B familial classic and his mutation analysis found that he had inherited a high risk allele (NF2CA3 allele "1") with no mutations. This indicates mild to moderate severity of disease [5]. The score classifies 2A as a full or mosaic NF2 mutation while 2B can indicate truncating mutations (associated with increased morbidity and mortality).
He was known to have a number of intracranial and spinal lesions typical of the condition (Figure 1) including: Bilateral vestibular schwannomas, bilateral trigeminal nerve schwannomas, a left sided oculomotor nerve schwannoma, a right sided facial nerve schwannoma, multiple intracranial meningiomas, multiple spinal cord ependymomas, a thoracic spinal meningioma and multiple intra- and extra-dural spinal schwannomas.
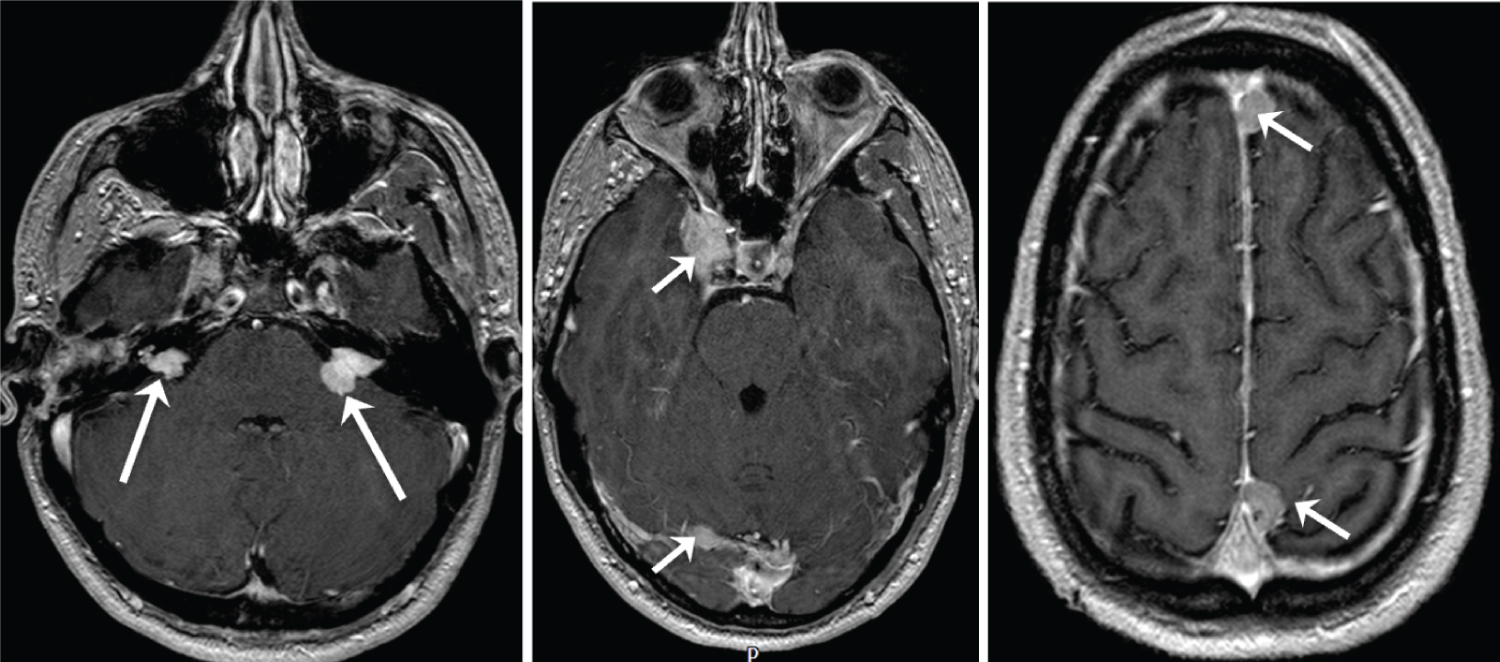 Figure 1: Selected axial gadolinium enhanced, T1 weighted MR images of the head performed in March 2020 demonstrating bilateral vestibular schwannomas (long arrows) and a number of meningiomas (short arrows), including a right cavernous sinus meningioma. View Figure 1
Figure 1: Selected axial gadolinium enhanced, T1 weighted MR images of the head performed in March 2020 demonstrating bilateral vestibular schwannomas (long arrows) and a number of meningiomas (short arrows), including a right cavernous sinus meningioma. View Figure 1
On this presentation, the patient presented to John Radcliffe Hospital (Oxford) with right sided hip pain, anterior thigh pain and weakness in his legs that developed over 7 months, affecting his mobility. There was no history of preceding trauma, radiotherapy or steroid use. Examination revealed tenderness over the greater trochanter on palpation of the right hip with slight swelling however no overlying skin changes, signs of infection or masses were present.
Neurological examination was unchanged with intact reflexes and long standing reduced sensation in the right leg.
The patient had been diagnosed radiologically as having a longstanding right L4 nerve sheath tumour consistent on MRI and PET as a schwannoma. This had been managed conservatively due to its lack of significant growth on MRI (Figure 2), concerning radiological features on MRI and PET or associated problematic clinical symptoms. Review of a positron emission tomography - computed tomography (PET-CT) scan (Figure 3) performed 8 years earlier showed that the right L4 nerve-sheath tumour was not flurodeoxyglucose (FDG) avid at the time, which in addition to its very slow growth on MRI imaging, further indicated that it was a benign schwannoma at that time.
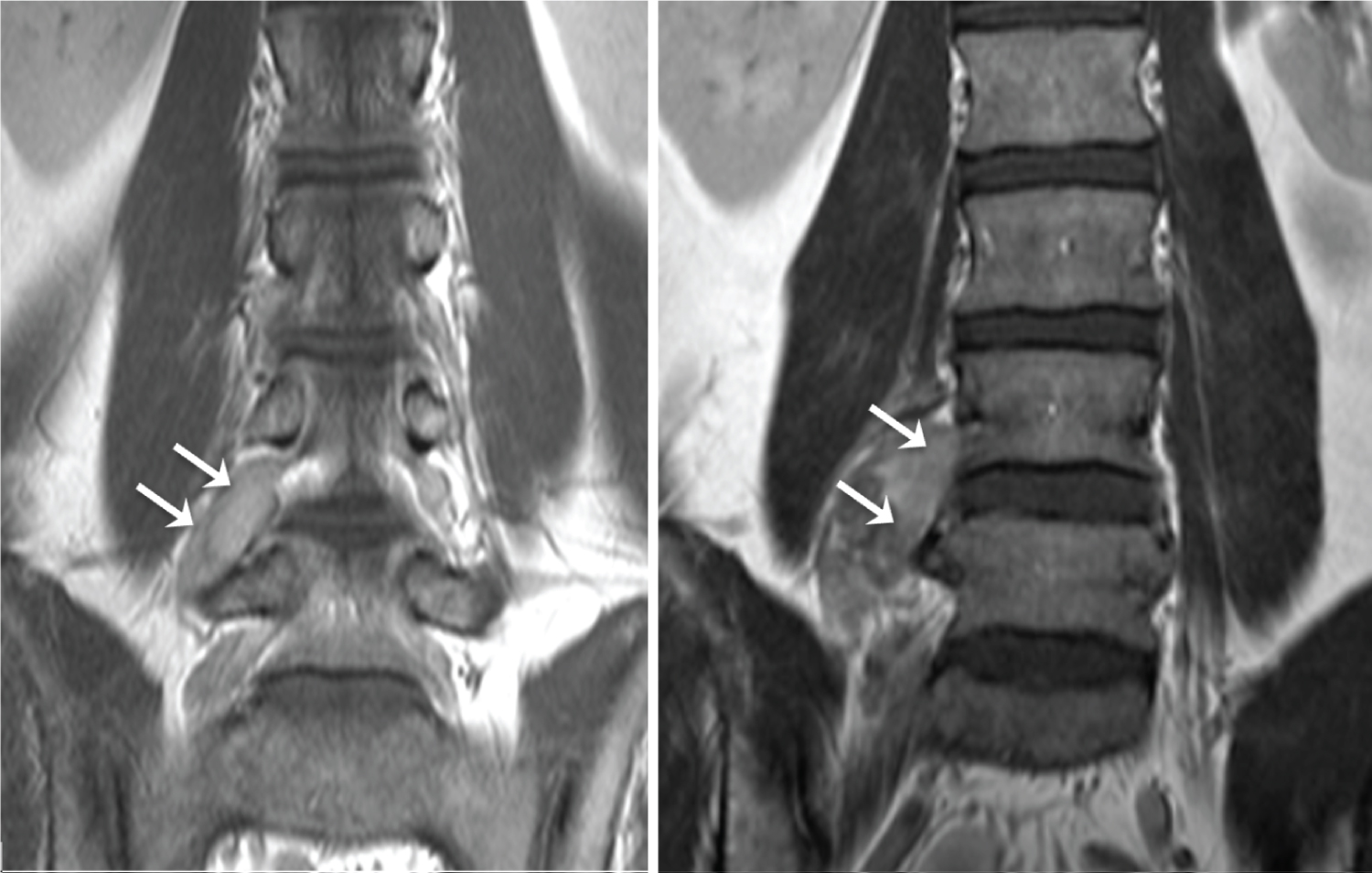 Figure 2: Coronal gadolinium enhanced T1 weighted images of the lumbar spine performed in June 2009 (left) and March 2020 (right). The right L4 nerve root schwanomma (arrows) show a relatively modest increase in size. View Figure 2
Figure 2: Coronal gadolinium enhanced T1 weighted images of the lumbar spine performed in June 2009 (left) and March 2020 (right). The right L4 nerve root schwanomma (arrows) show a relatively modest increase in size. View Figure 2
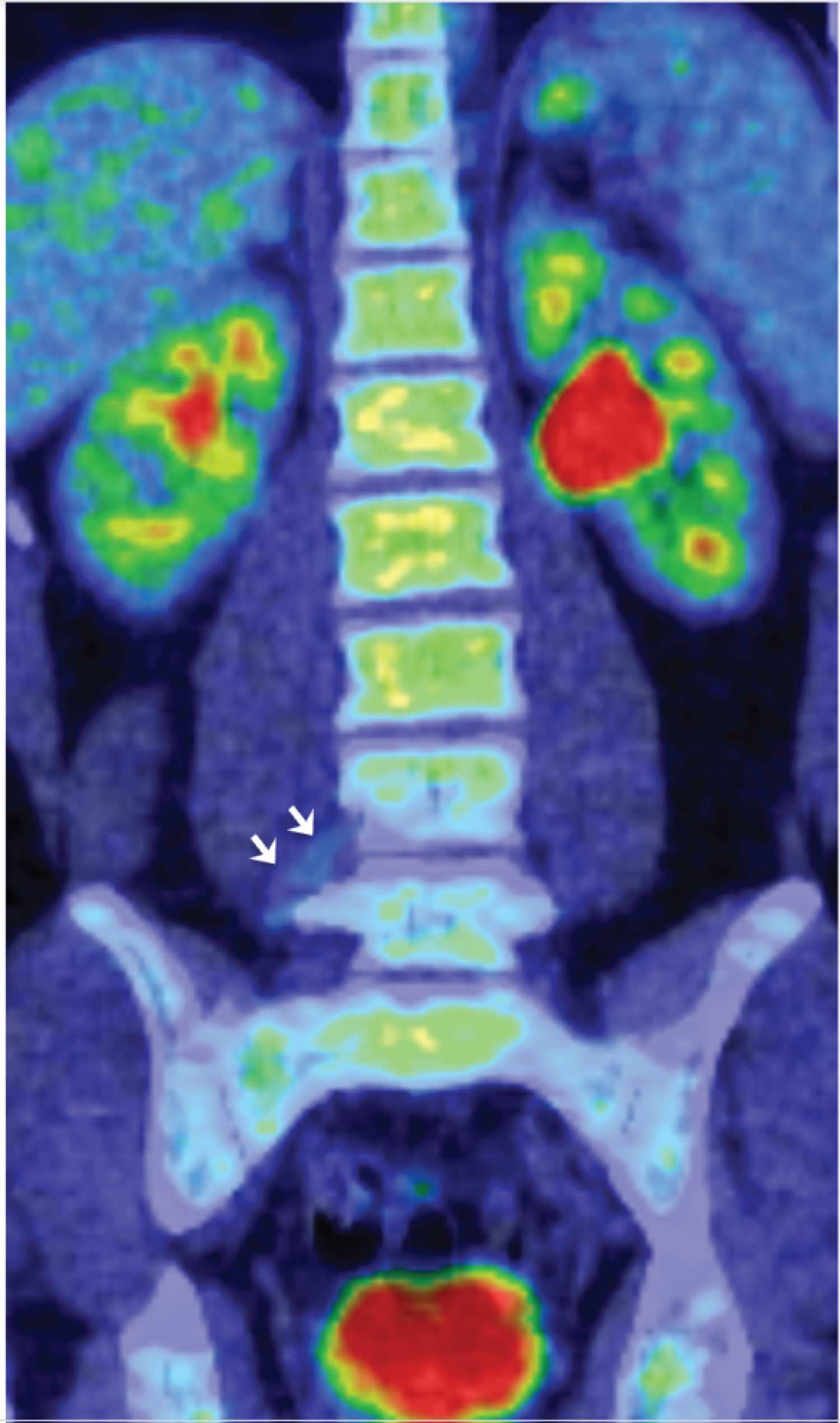 Figure 3: Coronal reconstruction of the 18 FDG PET-CT scan performed in 2012. The right L4 nerve root tumour is barely visible on this scan (arrows) since it is not metabolically active, and therefore not FDG avid. This is in keeping with a benign schwannoma at the time. View Figure 3
Figure 3: Coronal reconstruction of the 18 FDG PET-CT scan performed in 2012. The right L4 nerve root tumour is barely visible on this scan (arrows) since it is not metabolically active, and therefore not FDG avid. This is in keeping with a benign schwannoma at the time. View Figure 3
Due to the change in his symptoms, a magnetic resonance imaging (MRI) scan of the spine (Figure 4) was arranged. This showed that the right L4 nerve-sheath tumour (diagnosed radiologically as schwannoma) had increased in size dramatically since the previous MRI scan 8 months earlier. In its maximum coronal measurements this tumour had increased from 3.7 cm to 4.3 cm in an eleven year period leading up to the onset of the symptoms, it then increased to 13.5 cm over the next 8 months which captured the period in which he developed the above described symptoms. The signal characteristics of the lesion had also become more heterogeneous over that period. In addition to the rapid tumour growth, the scan also showed multiple bony metastases in the spine with pathological compression fractures of T4 and L3. These features were highly suspicious for malignant transformation of the L4 schwannoma. In light of these concerning MRI changes a CT scan of the chest was arranged. This unfortunately demonstrated multiple lung and bony metastases, and a malignant pleural effusion.
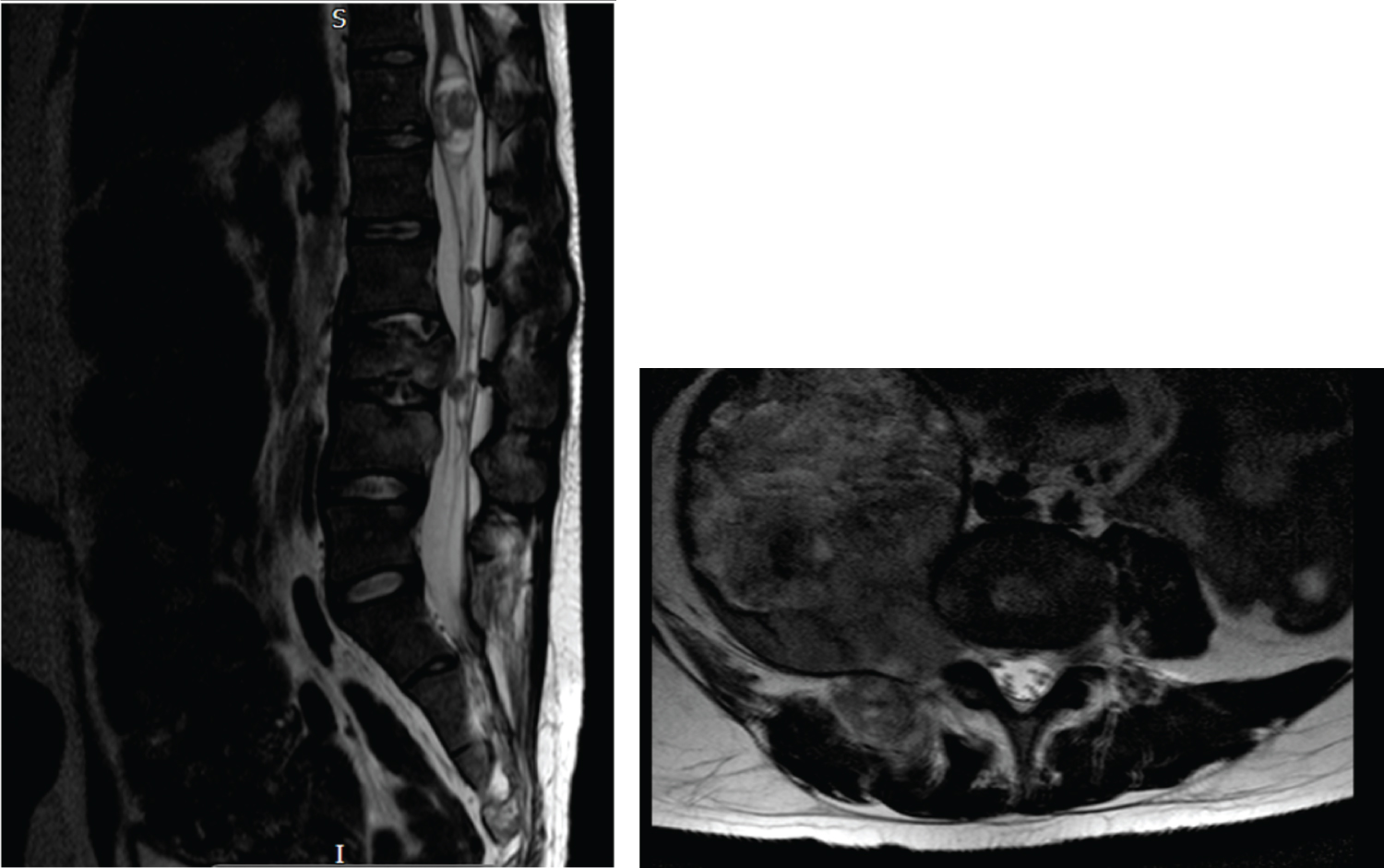 Figure 4: Sagittal (left) and coronal (right) STIR images performed in November 2020. Extensive patchy marrow infiltration throughout the spine (long arrows) with restricted diffusion consistent with cellular infiltration. The large extraforaminal mass from the L4 nerve root (short arrow) demonstrates heterogenous diffusion characteristics with slightly increased infiltration of the right pedicle and facet of L4 vertebra by soft tissue.
View Figure 4
Figure 4: Sagittal (left) and coronal (right) STIR images performed in November 2020. Extensive patchy marrow infiltration throughout the spine (long arrows) with restricted diffusion consistent with cellular infiltration. The large extraforaminal mass from the L4 nerve root (short arrow) demonstrates heterogenous diffusion characteristics with slightly increased infiltration of the right pedicle and facet of L4 vertebra by soft tissue.
View Figure 4
An ultrasound guided biopsy of the L4 lesion was urgently performed which confirmed malignant transformation of a peripheral nerve sheath tumour (MPNST). The histology report (Figure 5) concluded that: the retroperitoneal mass biopsy contained a very small amount of tumour fragments showing malignant features with very high proliferation index, myxoid changes and loss of INI1. The tumour was moderately cellular and displayed a myxoid background. The tumour cells displayed basaloid oval/elongated nuclei with fairly dense chromatin and some distinct nucleoli. The cytoplasm was indistinct and wispy, merging with the pale myxoid matrix. No definite mitotic figure was identified. Given the constellation of findings, the differential diagnoses of MPNST and other INI1-deficient tumours/sarcomas were considered likely but definitive diagnosis from this biopsy was not possible.
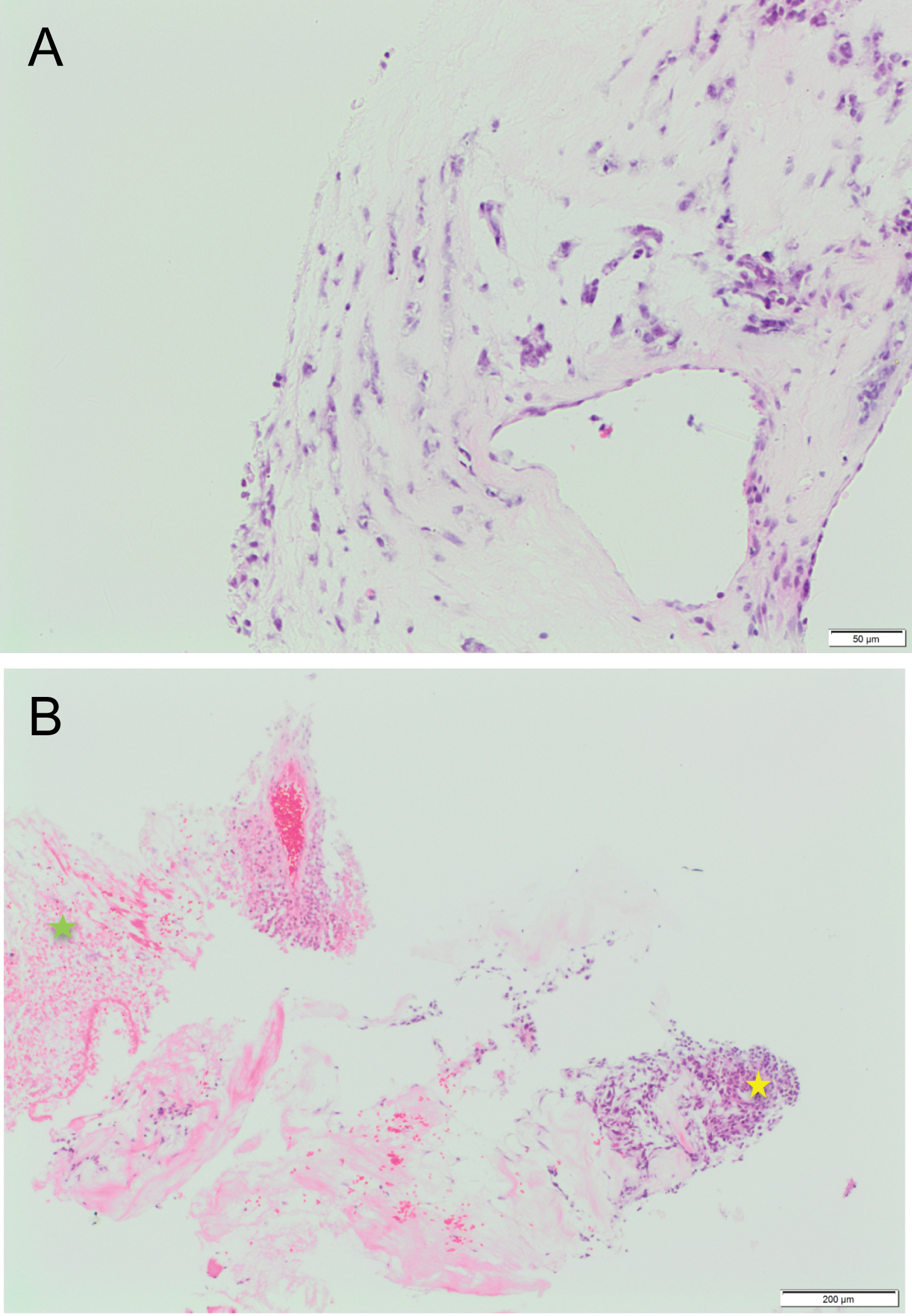 Figure 5a: H&E stained section demonstrating myxoid areas of tumour with moderate cellularity. The cytoplasm is indistinct and wispy, merging with the pale myxoid matrix.
Figure 5a: H&E stained section demonstrating myxoid areas of tumour with moderate cellularity. The cytoplasm is indistinct and wispy, merging with the pale myxoid matrix.
Figure 5b: H&E stained section showing more cellular component of the tumour (yellow star) composed of basaloid oval/elongated nuclei with dense chromatin. Tumour cell necrosis in other areas (green star).
View Figure 5
While the patient was being investigated for this new malignancy, he desaturated and a CTPA was then carried out initially querying a PE but found new nodularity in the posterior segment of the right upper lobe concerning for pulmonary metastasis as well as evidence of consolidation consistent with pneumonia. Accordingly, he was started on treatment for a hospital acquired pneumonia and urgent MDT discussions occured.
Options for management were carefully considered at our sarcoma MDT. Due to his widespread metastases surgical interventions were not considered appropriate. Information from further biopsy was also not considered necessary at that stage as treatment options would not have been altered by this. As such he was taken under the care of the oncology team after discussion at the sarcoma MDT. He underwent radiotherapy for cord compression at T4-T5 (8Gy single fraction) and to the right psoas mass and lumbar spine 30Gy in ten fractions, for 2 weeks. He also completed one cycle of infusional ifosfamide chemotherapy. Unfortunately while undergoing this treatment he developed signs of sepsis, was diagnosed with atypical pneumonia and was treated again with intravenous antibiotics. A further CT scan demonstrated disease progression of the pulmonary masses. Unfortunately due to continuing generalised sepsis despite antibiotic therapy ongoing cycles of chemotherapy could not be continued.
The difficult decision was made to palliate the patient and provide symptom control with the aim to discharge the patient home for end of life care. Sadly he subsequently continued to rapidly deteriorate and passed away approximately 6 weeks after diagnosis.
This case highlights a patient with NF2 who had undergone an extremely rare malignant transformation of a spinal nerve root schwannoma.
MPNST, also known as neurofibrosarcomas and malignant schwannomas, are a rare type of soft tissue sarcoma, thought to be of neural crest origin, typically associated with NF1 (50% of cases are associated with NF1, and 50% are sporadic) [6] rather than NF2 [7]. Only 5% of those with NF1 develop MPNST, making it a rare diagnosis [8] even in those with NF1. It is not known to be associated with NF2 (i.e. there is not an increased incidence in this group despite their relatively high burden of schwannomas). Spinal MPNSTs are particularly rare and associated with worse outcomes when compared with cellular schwannomas of the spinal canal [9]. Incidence is between 20 to 50 years with sporadic tumours occurring in the latter half.
MPNSTs are frequently misdiagnosed as they can present with variable neurology. They can present with pain, paraesthesia and motor weakness as it enlarges. Both radiologic and immunohistochemical studies are required to confirm the diagnosis [10] as imaging may be non-specific. Imaging studies that demonstrate rapid growth in a previously slow-growing tumour are of particular concern, as is evidence of invasion of adjacent structures, including bone invasion - as opposed to bony remodelling. The patient we describe had an initially slow growing mass over 11 years and then rapid growth was noted in the last 8 months of his latest presentation, involving the vertebra with signal changes that were consistent with malignant transformation. A study [11] looking at radiological features of MPNSTs described features such as tumour size greater than 5 cm, ill defined margins, peritumoural oedema and heterogeneous enhancement as aiding diagnosis. In our patient the size of the lesion was greater than 5 cm and there was heterogeneity of the internal signal and enhancement characteristics, but in our experience larger schwannomas in patients with NF2 can often display those imaging characteristics. The MRI scan demonstrating rapid growth, combined with invasion into surrounding soft tissues and bone as well as vertebral metastases at other levels were however a strong indication of malignant change in the tumour.
Diagnosing this extremely rare condition in a patient with NF2 is challenging due to the large number and marked variation in growth rate of benign schwannomas seen in this group of patients, combined with the heterogeneity in imaging characteristics.
Immunohistochemical analysis of MPNST normally shows elongated nuclei and perivascular hypercellularity. In our patient we were not able to attain sufficient samples, however findings of an elongated nucleus made malignant transformation likely.
Surgical resection forms the mainstay of treatment for MPNST [12] with addition of radiotherapy or chemotherapy. Though no standard regimen exists due to the rarity of the condition, those with low grade tumours will be more likely to undergo surgical resection while those with higher grade tumours undergo chemo or radiotherapy. Surgery is associated with the highest rate of survival (53% at 5 years) while those undergoing chemo or radiotherapy alone had a 5 year survival rate of 42%. Our patient underwent chemotherapy for his MPNST as he was unable to undergo surgical resection due to widespread disease. Though this data is not specific to patients with a diagnosis of NF2, the rare malignant transformation of a PNST and the fact that only chemotherapy could be administered as treatment in our patient, may explain the unfortunate outcome.
A study [13] looking at 1253 patients diagnosed with NF2 found that there was no evidence that spontaneous MPNST was a feature of NF2. There were no cases of spontaneous MPNST reported and only 2 patients were reported to develop MPNST. Both patients either underwent stereotactic radiosurgery or were presumed to undergo radiotherapy for a vestibular schwannoma, 3 years prior to MPNST being diagnosed and it was thought that this may be the cause for the transformation. Our patient underwent radiotherapy after malignant transformation was noted, meaning he had no risk factors to undergo malignant change of his L4 nerve root lesion. The study mentioned was specifically looking at transformation seen in vestibular schwannoma while the case we have highlighted reports transformation in a spinal schwannoma and so it may not be appropriate to correlate results from this study to our case.
A case reported [14] in 2003 described multiple MPNST in a patient with NF2, however they did not confirm whether these were spontaneous or whether benign schwannomas had undergone malignant transformation. Of note was that the lesions in that case were both cranial and peripheral, while in our patient, only one of the lesions, which was a spinal schwannoma underwent malignant change, the other schwannomas remained grossly unchanged. Other studies [15,16] have linked NF1 with MPNST, finding a lifetime risk of 8-13%. Patients with NF1 were also found to develop MPNST at a much younger age, compared to those that developed spontaneous MPNST. A link with NF2 was not established, making the case we present, particularly rare.
In a study [17] following 293 patients, MPNST was reported in only 2% of those with spinal schwannomas, operated over a 38 year period. This study did not specify whether patients were diagnosed with NF2 or not, so the association between NF2 and MPNST cannot be determined from that study. The fact that incidence of MPNST is so rare, particularly those affecting spinal nerves (other studies have focused on malignant transformation in vestibular schwannomas) [18,19], also makes it difficult to analyse large enough sample sizes in order to determine causal factors. In the case we have described, no obvious risk factors can be identified to attribute to the malignant transformation seen.
The genetic severity score [5] in the patient we described was 2A/B indicating mild to moderate severity of disease. This is associated with an average life expectancy of 45.9 years. Predictors of disease severity include: The age of onset of symptoms and age at diagnosis, correlating with the growth rate of tumours and mortality. Presence of intracranial meningiomas is also associated with early mortality. Those in moderate to severe groups (groups 2 and 3) were more likely to have intracranial meningiomas, spinal meningiomas and schwannomas, increasing the tumour burden and contributing to poor prognosis. Our patient was diagnosed at 9 years of age and had a significant tumour burden, with intracranial and spinal meningiomas as well as spinal schwannoma. Our patient had moderate severity of disease with risk factors (early age and increased tumour load), however his age at death (30 years) was much younger than the associated life expectancy of his severity of disease, due to the rare malignant transformation of his peripheral nerve sheath tumour.
Malignant transformation of a PNST is reported very rarely in the NF2 setting. They carry high mortality and morbidity and for this reason, awareness of such cases is important to aid early detection.
The case highlighted demonstrates a rare incidence of transformation of a spinal nerve root schwannoma in a patient with NF2.
We thank Dr M Hofer (Department of Neuropathology, John Radcliffe Hospital, Oxford) for providing us with the immunohistochemical analysis and the pathology slides used in this report.
None.
No ethical approval is required for this case report.