Atypical teratoid/rhabdoid tumor (AT/RT) is a Grade 4 embryonal tumor, rare and aggressive, most often presenting in children 0-5 years of age. In the adult population, AT/RTs are exceedingly rare, and usually located intracranially in the supratentorial compartment. AT/RTs of the spine are even more uncommon. Here, we detail the case of a young adult with AT/RT of the lumbar spine who underwent surgical resection of the lesion followed by radiation and chemotherapy. We also provide a comprehensive review of reported cases of adult spinal AT/RT to date, and review the current literature on clinical management of adult AT/RT.
Atypical teratoid/Rhabdoid tumor, Spinal oncology, Clinical pathology
Atypical teratoid/rhabdoid tumor (AT/RT) is a rare and aggressive neoplasm of the central nervous system (CNS), categorized as a Grade 4 embryonal tumor by the World Health Organization [1,2]. Most often presenting in children between 0 and 5 years of age, AT/RT represents 1.6% of all pediatric CNS tumors, with the majority occurring in the posterior fossa at the cerebellopontine angle [3,4]. AT/RT are exceedingly less common in the adult population (≥ 18 years of age), with a recent study identifying 96 cases of adult ATRT reported in the literature [5-7]. The most common location of AT/RT in adults is found intracranially in the supratentorial compartment, with few reports of spinal AT/RT. Here, we describe a rare adult case of spinal AT/RT from our institution. Additionally we summarize the reported cases of adult spinal AT/RT to date (Table 1 [8-16]), and review the current literature on their management.
Table 1: Literature review of reported adult spinal AT/RT cases. View Table 1
A 26-year-old male with no significant past medical history presented with lower back pain, progressive lower extremity weakness with paresthesias and difficulty voiding over a period of several months. Magnetic resonance imaging (MRI) of the lumbar spine revealed an intradural extramedullary mass at the L4 level. Subsequent imaging of the neural axis revealed extensive abnormal surface enhancement of lower thoracic cord, a small linear focus along posterior margin of the cord at C3-4 disc space, and a foci of enhancement within the posterior fossa (Figure 1). These findings were concerning for leptomeningeal metastatic disease. The patient underwent surgical intervention with L4 laminectomy and L3 partial laminectomy for open biopsy and resection of the lumbar spine lesion, with resection of a 2 × 5 cm cauda equina mass. Intraoperatively, the cauda equina roots were notably expansile and matted together due to tumor infiltration. There was additional metastasis noted throughout the lumbar spine within the subarachnoid space as well as nerve roots. Final pathology described an INI-1 deficient malignant neoplasm with an immunophenotype suggestive of AT/RT.
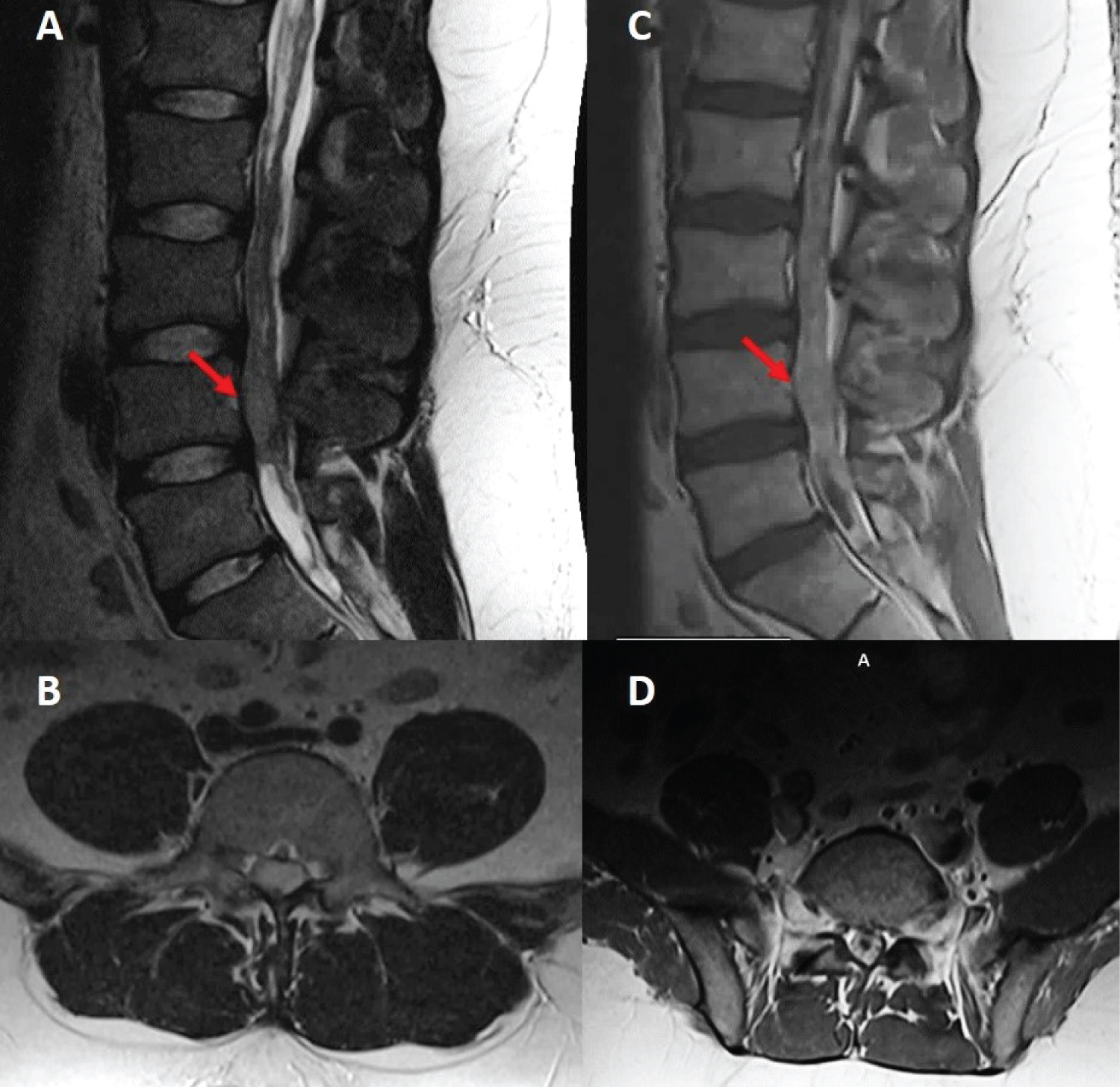 Figure 1: Sagittal (A) and axial (B) T2, sagittal (C) and axial (D) T1 post-contrast MRI sequences demonstrating an intradural and extramedullary lesion centered at the L4 level of the lumbar spine (red arrows).
View Figure 1
Figure 1: Sagittal (A) and axial (B) T2, sagittal (C) and axial (D) T1 post-contrast MRI sequences demonstrating an intradural and extramedullary lesion centered at the L4 level of the lumbar spine (red arrows).
View Figure 1
Within the first month postoperatively, the patient developed positional headaches and vision changes, especially in the left eye. Evaluation per neuro-ophthalmology identified papilledema, retinal hemorrhage, and worsening left eye exotropia. Repeat imaging revealed marked progression of thoracic and lumbar disease with no change in cervical or medullary foci (Figure 2). Spinal enhancement filled the dural space, obscuring the nerve roots and the tip of conus medullaris. Additionally, an intrathecal arachnoid cyst was noted to compress the spinal cord from T4 to T8, resulting in edema from T8-T11.
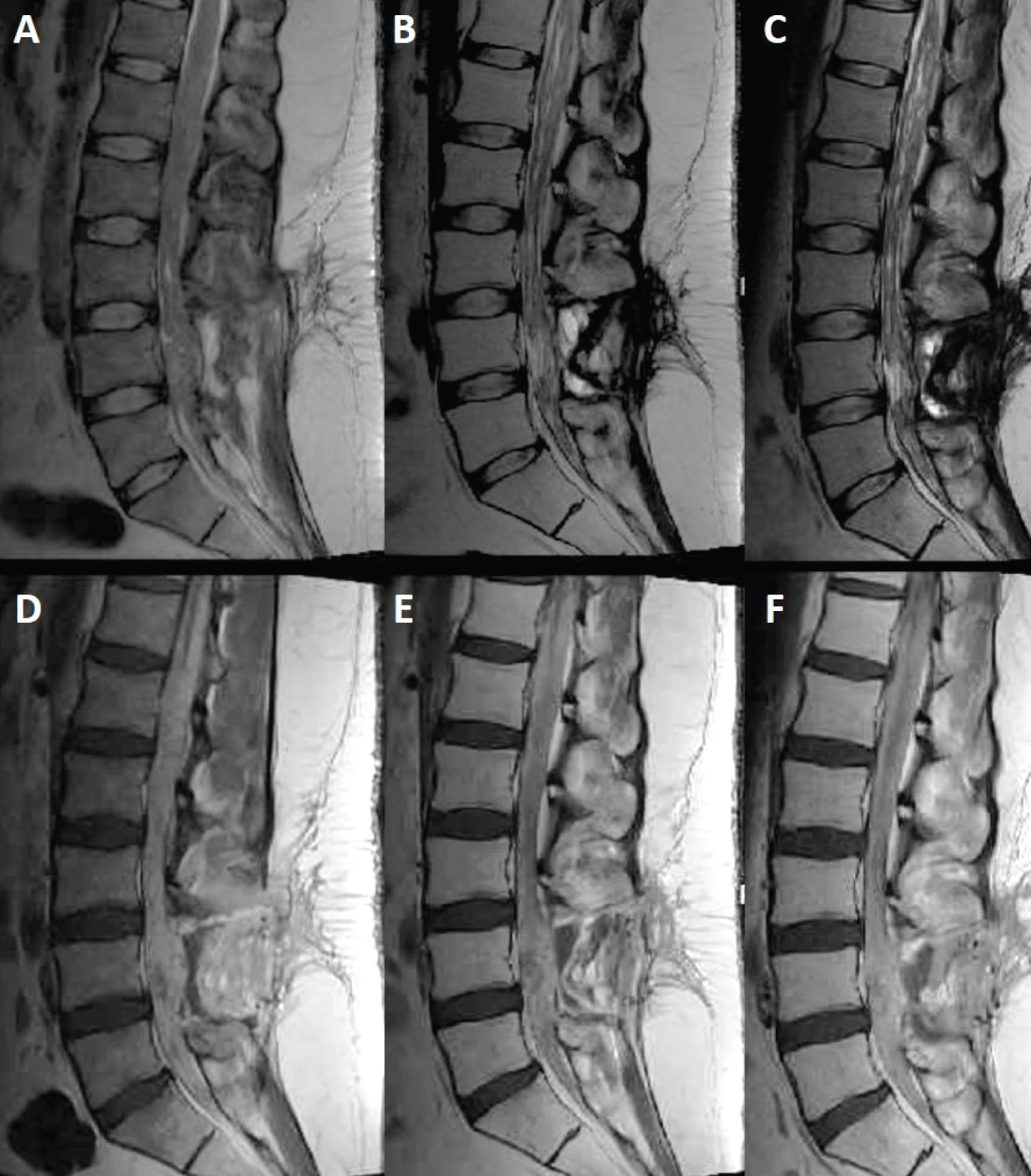 Figure 2: Sagittal T2 (top row) and T1 post-contrast (bottom row) MRI sequences demonstrating lesion progression at 1 month (A,D), 4 months (B,E) and 6 months (C,F) post-operatively.
View Figure 2
Figure 2: Sagittal T2 (top row) and T1 post-contrast (bottom row) MRI sequences demonstrating lesion progression at 1 month (A,D), 4 months (B,E) and 6 months (C,F) post-operatively.
View Figure 2
In accordance with the medulloblastoma treatment protocol, the patient underwent craniospinal irradiation followed by multi-agent chemotherapy (cyclophosphamide/vincristine/cisplatin). His treatment course was complicated by neutropenic fever, saddle pulmonary embolism requiring IVC filter placement, acute kidney injury, uremic encephalopathy necessitating hemodialysis, recurrent upper gastrointestinal bleeding and cardiac arrest. He received only C1 of chemotherapy, with treatment being discontinued due to significant complications from the treatment.
Less than one year post-resection, after completing radiation and partial chemotherapy, MRI brain and spine were stable through serial checks. However, the patient continued to endorse left-sided headache with worsening exotropia, accompanied by urinary incontinence and dramatic weight loss. He was found to have acute metabolic encephalopathy and communicating hydrocephalus, requiring palliative left ventriculoperitoneal shunt placement. Despite aggressive measures, he continued to decline and passed away approximately twelve months after diagnosis.
Histologic examination revealed hypercellular embryonal neoplasm composed of small cells with intermittent nucleoli and high mitotic activity on a somewhat hyalinized background matrix (Figure 3A).
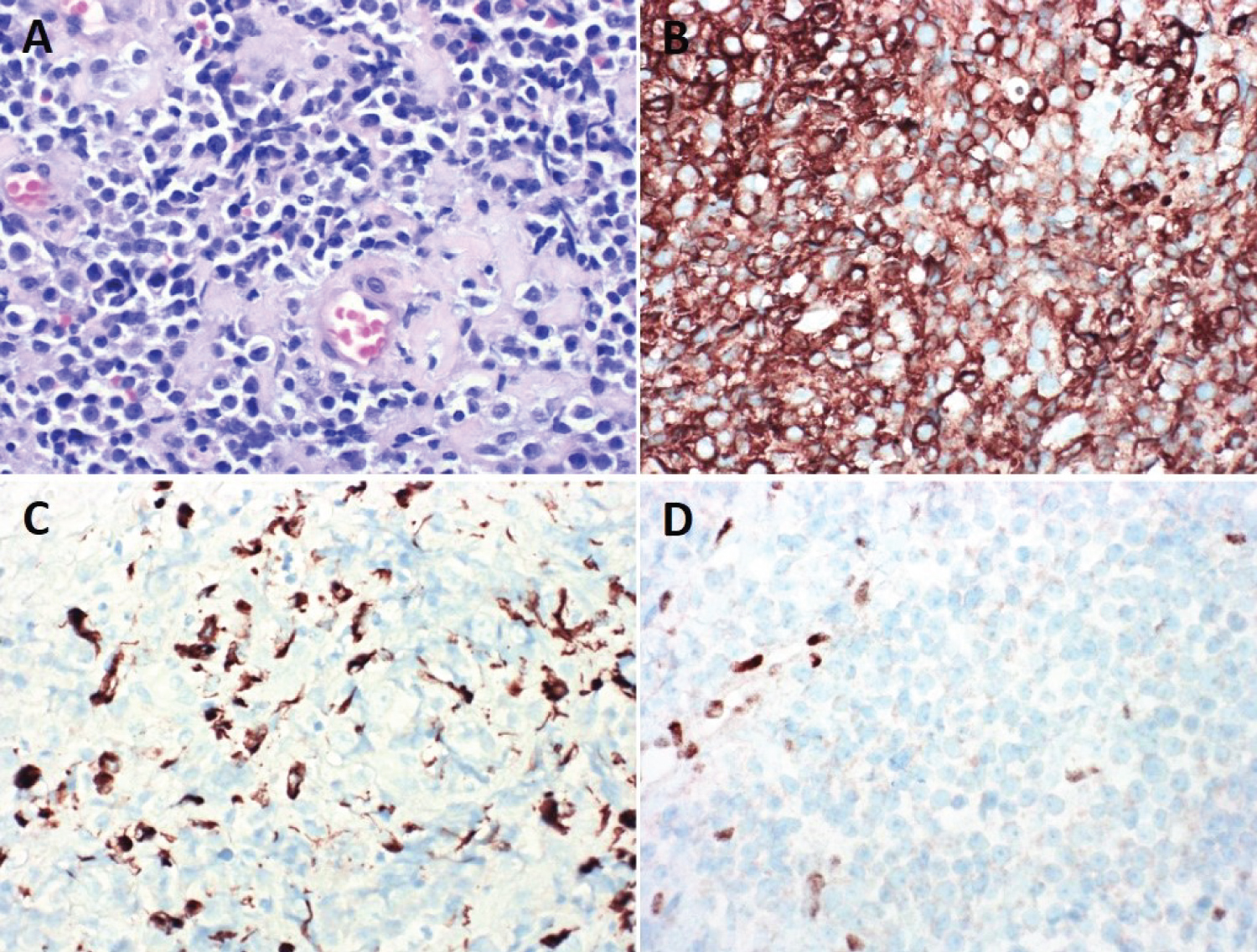 Figure 3: A) Microscopic and immunohistocytochemistry pathology analysis of the tumor sample (40x magnification) - H&E; B) Smooth muscle actin; C) Cytokeratin CAM5.2; D) INI1.
View Figure 3
Figure 3: A) Microscopic and immunohistocytochemistry pathology analysis of the tumor sample (40x magnification) - H&E; B) Smooth muscle actin; C) Cytokeratin CAM5.2; D) INI1.
View Figure 3
Tumor cells demonstrated immunopositivity for smooth muscle actin (SMA) (Figure 3B), cytokeratin CAM 5.2 (Figure 3C), and epithelial membrane antigen (EMA). WT-1 immunoreactivity was present in tumor nuclei. Tumor cells were immunonegative for glial (Olig-2, GFAP), neuronal (synaptophysin, S-100), and muscle (myogenin, myoD1, desmin) markers. Immunohistochemical staining demonstrated loss of INI-1 in tumor cells with retained expression in stromal elements (Figure 3D). Subsequent fluorescence in situ hybridization (FISH) testing demonstrated 22q/INI1 deletion. Other INI-1 losing tumors were considered including myoepithelial carcinoma and epithelioid sarcoma. However, given the unique immunophenotype along with the intradural tumor location, the best classification was felt to be that of AT/RT.
AT/RT is an exceedingly rare and aggressive malignant neoplasm, presenting both a diagnostic and therapeutic challenge for clinicians [2]. While in the pediatric population AT/RTs account for 4.4% of CNS tumors in ages 0-5 years, in adults the diagnosis of AT/RT is uncommon [17]. A recent study by Broggi, et al. identified 96 reported cases of adult AT/RT in the literature [7]. The majority of patients were female (72.9%) and had tumors located supratentorially in 81.3% of cases [7]. Only 7.3% were found to involve the spinal cord. To our knowledge, we present the ninth case ever reported of spinal AT/RT in adult patients. Histologically, AT/RT often present sheets of rhabdoid cells may be predominant, but may in other instances exhibit primitive neuroectoderm-like phenotype, which may be difficult to differentiate from other embryonal tumors such as medulloblastoma [3,18,19]. Radiographically, spinal AT/RT is generally heterogenous and often mistaken for more common tumors of the spine such as nerve sheath tumors or chordomas [20,21]. Furthermore, tumors extending through neural foramina may mimic schwannoma, malignant peripheral nerve sheath tumors, or even neurofibromatosis [20-22].
Given that AT/RT may be ambiguous on imaging or histology alone, tumor markers and immunohistochemical analysis play an essential role. AT/RT were first recognized by their polyimmunophenotype, specifically expression of both mesenchymal markers such as smooth muscle actin and epithelial markers such as cytokeratin or epithelial membrane antigen. More definitive diagnosis is possible now using immunostaining for INI-1, as loss of INI-1 nuclear staining indicates biallelic inactivation of SMARCB1 on chromosome 22 [19,23]. Although rarely AT/RT may have biallelic inactivation of the alternative SWI/SNF complex genes such as SMARCA4/BRG1, inactivation of SMARCB1/INI1 remains the most consistent and definitive diagnostic tool available [24-26]. While the exact role of INI-1 in AT/RT pathogenesis is unknown, its inactivation likely alters cell cycle activity to promote tumorigenesis [26]. Additionally, recent studies have identified genetic variations via whole exome sequencing that offer insight into the specific manner in which SMARCB1 is inactivated and the possibility of definitive diagnosis [5,27,28]. Among the cases of adult spinal AT/RT reported (Table 1), those that performed immunostaining for INI-1 observed negative reactivity. Additionally, two of the cases identified 22q deletions via FISH.
The diagnostic challenge, high incidence of local recurrence, and propensity for leptomeningeal dissemination collectively contribute to a poor prognosis of AT/RT [29]. Without an identified biological prognostic factor to explain long-term survival in AT/RT, recent studies have aimed to seek explanation elsewhere [30]. Adults appear to have a better prognosis than pediatric patients. This is likely due to tumor location, which is often supratentorial in adults. Also, adult patients can tolerate more aggressive treatment, such as craniospinal radiation, which is effective for AT/RT, but often cannot be tolerated by pediatric patients [31]. Prompt resection followed by focal radiotherapy and multi-agent chemotherapy has been found to prolong survival in pediatric AT/RT [32]. Similarly, in the largest single center series of pediatric primary spinal AT/RT, patients with progression-free survival > 1 year all received multimodal therapy with or without intrathecal chemotherapy, focal radiation, or myeloablative chemotherapy [33]. Robust data for adult-onset AT/RT is lacking due to the rarity of the condition on this patient population. In the most recent systematic review of adult AT/RTs, the mean survival after treatment was 15 months [7]. Combined treatment with surgical resection, chemotherapy and radiation showed a significantly higher survival benefit than combined chemotherapy and radiation without surgery, and especially than simply chemotherapy or radiotherapy alone [7]. Additionally, in adult AT/RT cases with survival > 3 years, tumor location was found to be predominantly hemispheric [34].
Among the ten cases of adult spinal AT/RT described, including the present case, the mean age at diagnosis was 33.9 years, ranging from 19 to 65 years of age, as shown in Table 1. The ratio of female to male was 1:1. 67% of patients presented with lesions in the lower thoracolumbar spine, while 33% of patients had lesions in the cervical spine. Leptomeningeal spread was present in 80% of cases where presence or absence of leptomeningeal disease was noted. The most common therapy included surgical resection followed by radiation (50%), followed by surgical resection with combined chemotherapy and radiation (38%) [11,13]. One patient was treated with combined chemotherapy and radiation without surgical resection. There were no patients treated with surgery, chemotherapy or radiotherapy alone. Of those patients who underwent surgery, gross total resection was achieved in only 43% of patients. The average survival was 17 months, ranging between 6 months up to 3 years at last follow up. Additionally, the majority of cases reported ≥ 1 relapse despite treatment.
Several chemotherapeutic regimens have been described for the treatment of AT/RT, including the modified Intergroup Rhabdomyosarcoma III protocol, St. Jude medulloblastoma protocol, malignant rhabdoid tumor protocol and more [11,13,35-38]. The majority of chemotherapy agents in these protocols include vincristine, cyclophosphamide, cisplatin, and etoposide. In the past several years, with the surge in molecular profiling and targeted therapies, multiple agents have emerged with multiple clinical trials in progress. Some of these agents target chromatin remodeling and histone modifications, especially histone deacetylase inhibitors such as panobinostat and EZH2 inhibitor tazemetostat [11]. Alisertib, an aurora A kinase inhibitor involved in cell cycle regulation is being tested in a phase II trial for recurrent of progressive AT/RT [32]. While there exist no guidelines for optimal management of adults with AT/RT, emerging treatment avenues in the pediatric population are important and could potentially be translated to adults. For instance, Rao, et al. used a modified ACOG ACNS 0332 regimen to treat an adult patient diagnosed with sellar AT/RT, which demonstrated decrease in tumor size in follow up imaging [39]. A similar strategy was used with the patient treated at our institution. In regards to spinal AT/RTs, resection is possible as the lesion appears to be intradural and extramedullary. The feasibility of maximal resection, however, will depend on an individual basis. Nevertheless, proper recognition of spinal AT/RT in adults is important to give appropriate prognostic information and to initiate aggressive treatments. As such, optimization of treatment regimen merits continued investigation.
In conclusion, spinal AT/RT in adults comprises a unique subset of tumor with minimal data guiding diagnosis and treatment. We describe a case of an adult patient presenting with cauda equina syndrome and neurogenic bladder secondary to spinal AT/RT with leptomeningeal spread. As the tenth case ever reported, this study aims to add to the paucity of literature on adult spinal AT/RT and expand the differential of rare spinal tumors in adults.
None.
None.