A 62-year-old woman with newly diagnosed systemic AL amyloidosis was evaluated for severely increased left ventricular (LV) wall thickness. Her echocardiographic, cardiac MRI, and genetic findings led to a diagnosis of hypertrophic cardiomyopathy (HCM). Five years after achieving relative a partial hematologic response with chemotherapy, the patient died after developing rapidly progressive heart failure and cardiogenic shock. A full autopsy diagnosed both cardiac amyloidosis and HCM. The overlapping phenotypes of these two diseases resulted in a clinical diagnostic dilemma. This case highlights the importance of tissue diagnosis and genetic analysis in the evaluation of LV wall thickening when conflicting clinical features suggest divergent diagnoses.
AL amyloidosis, Transthyretin, Hypertrophic cardiomyopathy
We present a case of increased left ventricular wall thickness in a patient with AL amyloidosis. The patient was ultimately diagnosed with both hypertrophic cardiomyopathy and AL cardiac amyloidosis. The overlapping phenotypes of these two diseases resulted in a clinical diagnostic dilemma. This case highlights the importance of tissue diagnosis and genetic analysis in the evaluation of LV wall thickening when conflicting clinical features suggest divergent diagnoses. In addition, clinicians should consider superimposition of AL amyloid cardiomyopathy in patient with rapid clinical deterioration.
A 62-year-old woman with hypertension and newly diagnosed AL amyloidosis was initially referred to the Boston University/Boston Medical Center Amyloidosis Center in 2006 for evaluation of suspected amyloidosis and increased left ventricular wall thickness noted by echocardiogram. There was no significant family history of heart disease or sudden cardiac death. A bladder biopsy obtained during evaluation for persistent hematuria revealed extensive amyloid disposition. A bone marrow biopsy xsmarrow biopsy. This was accompanied by markedly elevated serum lambda free light chains and monoclonal lambda protein in urine and serum immuno fixation electrophoresis, confirming the diagnosis of systemic AL amyloidosis (Table 1). Treatment with nine cycles of melphalan and dexamethasone resulted in a very good partial hematologic response with normalization of serum free light concentration, but persistent monoclonal protein in serum and urine by immunofixation electrophoresis and 5-10% lambda-restricted plasma cells on subsequent bone marrow biopsies.
Table 1: Hematological workup resulting in diagnosis of plasma cell dyscrasia. View Table 1
A screening transthoracic echo at presentation to our center showed a severely increased interventricular septal thickness of 22 mm, grade 1 diastolic dysfunction pattern, systolic anterior motion of the mitral valve with a left ventricular outflow gradient of 46 mmHg, mild mitral regurgitation, and left ventricular ejection fraction (LVEF) of 50% (Figure 1, Video 1A, Video 1B and Video 1C). Further testing demonstrated a serum Brain Natriuretic Peptide of 1388 pg/mL (normal < 53.2 pg/ml). An electrocardiogram showed normal sinus rhythm, left ventricular hypertrophy, left anterior fascicular block, and ST-T waves changes consistent with repolarization abnormality (Figure 2). Cardiac MRI (CMR) revealed a septal thickness of 19 mm and left ventricular mass of 532 g, LVEF of 59%, right ventricular ejection fraction of 52%, and a single focus of delayed enhancement within the anterior wall in a pattern that did not suggest an infiltrative cardiomyopathy (Figure 3 and Video 2). These CMR and echocardiographic findings confirmed a diagnosis of hypertrophic cardiomyopathy (HCM).
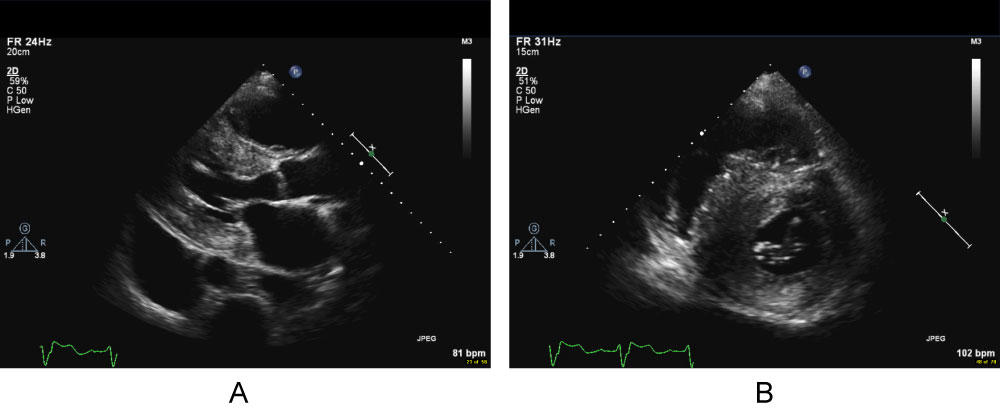 Figure 1: Cine cardiac MRI apical long axis movie illustrating systolic anterior motion of the mitral valve and flow acceleration in the left ventricular outflow tract.
View Figure 1
Figure 1: Cine cardiac MRI apical long axis movie illustrating systolic anterior motion of the mitral valve and flow acceleration in the left ventricular outflow tract.
View Figure 1
Video 1A: Video clips of the transthoracic echocardiogram (TTE) obtained during initial evaluation at the Boston University Amyloidosis Center. A) Parasternal long-axis;
Video 1B: B) Parasternal short-axis;
Video 1C: C) And apical four-chamber (C) Views reveal severely increased LV wall thickness with asymmetric septal hypertrophy, preserved LV systolic function and systolic anterior motion (SAM) of the anterior mitral valve leaflet.
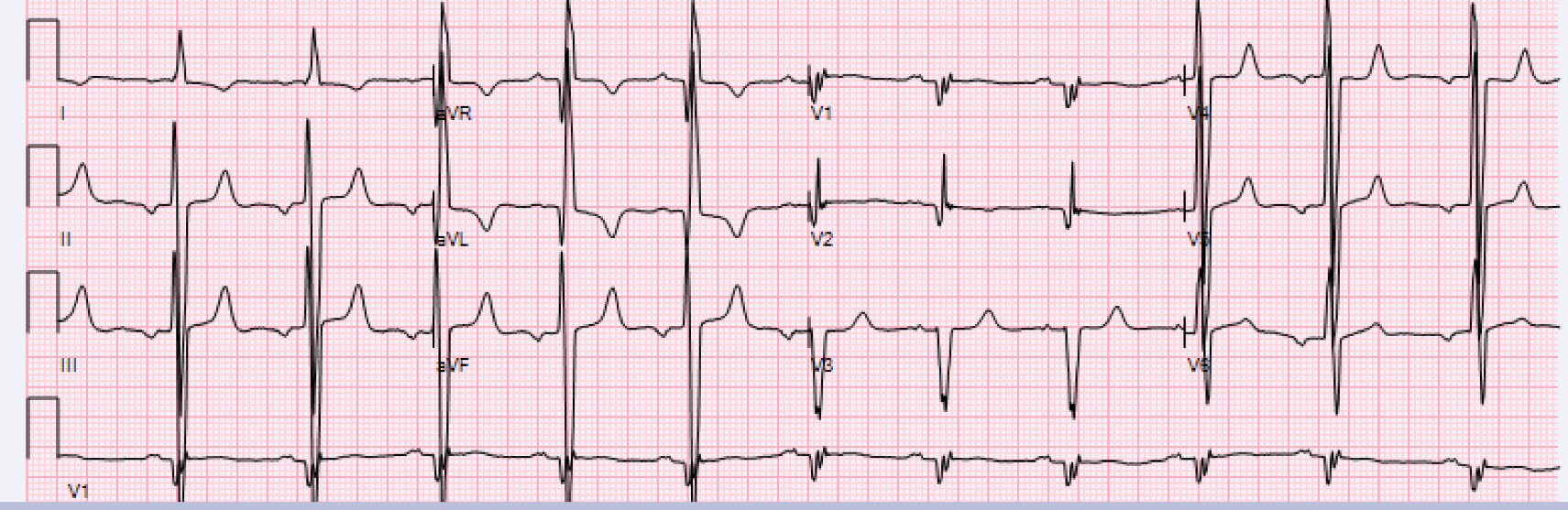 Figure 2: Routine ECG during Cardiology Clinic Visit. ECG reveals sinus rhythm, left ventricular hypertrophy, left anterior fascicular block, and ST-T waves changes consistent with repolarization abnormality.
View Figure 2
Figure 2: Routine ECG during Cardiology Clinic Visit. ECG reveals sinus rhythm, left ventricular hypertrophy, left anterior fascicular block, and ST-T waves changes consistent with repolarization abnormality.
View Figure 2
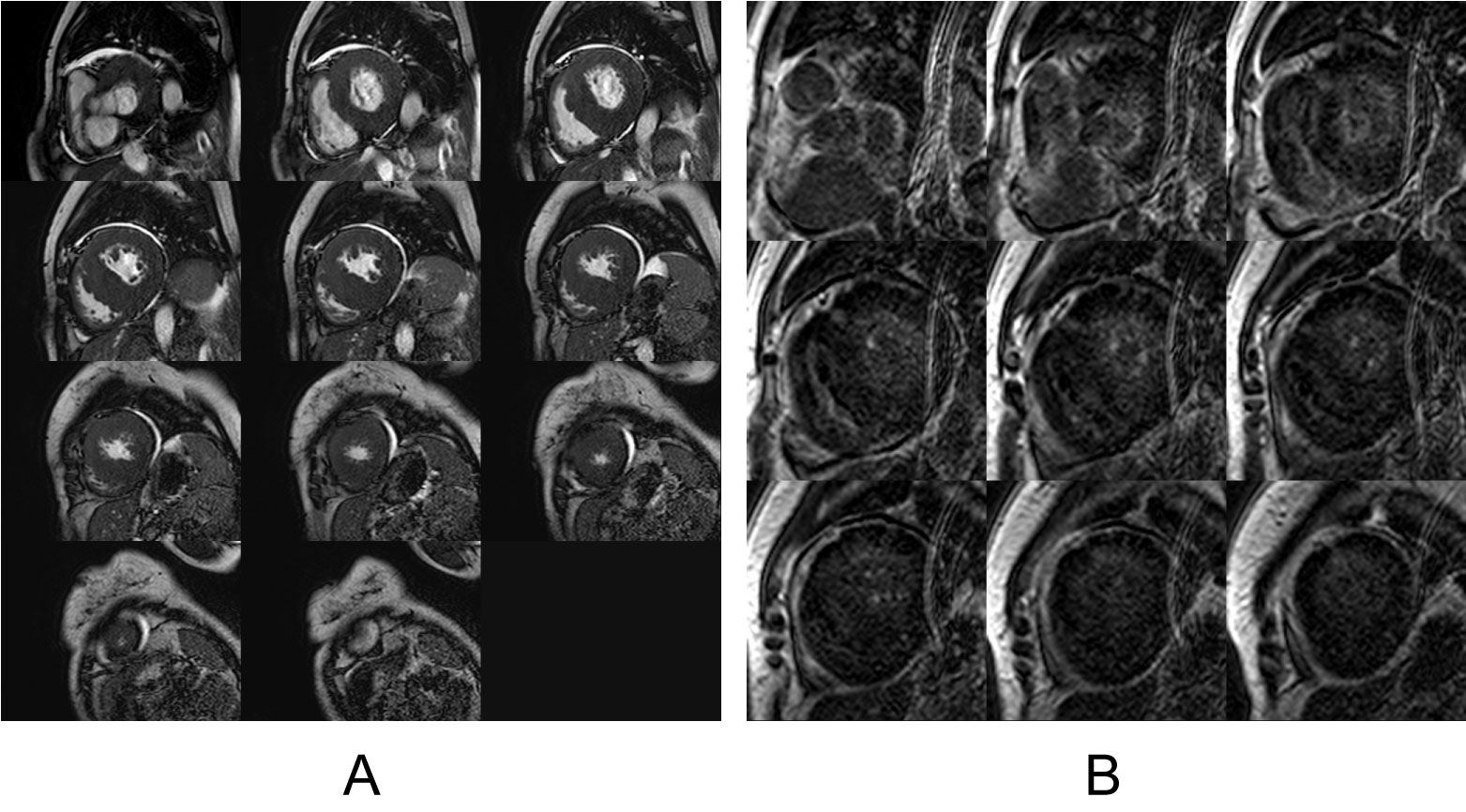 Figure 3: Cardiac MRI. A) Still-frame cine cardiac MRI contiguous short axis image slices from base to apex are illustrated. Note the prominent and asymmetric septal thickening consistent with HCM; B) Still frame late gadolinium enhancement (LGE) cardiac MRI short axis image slices from base to apex are illustrated. Note the focal late enhancement (single arrow) of the interventricular septum in a pattern consistent with HCM. Also note the poor contrast between myocardium and blood pool with subtle subdendocardial LGE (double arrow) that could also be consistent with cardiac amyloidosis.
View Figure 3
Figure 3: Cardiac MRI. A) Still-frame cine cardiac MRI contiguous short axis image slices from base to apex are illustrated. Note the prominent and asymmetric septal thickening consistent with HCM; B) Still frame late gadolinium enhancement (LGE) cardiac MRI short axis image slices from base to apex are illustrated. Note the focal late enhancement (single arrow) of the interventricular septum in a pattern consistent with HCM. Also note the poor contrast between myocardium and blood pool with subtle subdendocardial LGE (double arrow) that could also be consistent with cardiac amyloidosis.
View Figure 3
Video 2: Cine cardiac MRI apical long axis movie illustrating systolic anterior motion of the mitral valve and flow acceleration in the left ventricular outflow tract.
Blood testing for cardiac sarcomere mutations was sent and family members screened. There were no affected first-degree relatives, however the patient's genetic testing was significant for a missense mutation in MYH7, a β-myosin heavy chain gene, associated with HCM [1].
Over a five-year period, the patient slowly started to develop signs and symptoms of congestive heart failure (Table 2). The LVEF fluctuated, but largely remained preserved (Table 3). However, the septal wall thickness gradually began to increase and reached up to 29 mm.
Table 2: Clinical characteristics and laboratory data of the patient from diagnosis until death. View Table 2
Table 3: Echocardiographic characteristics of the patient from diagnosis until death. View Table 3
Six years after her initial presentation, the patient was acutely hospitalized for decompensated heart failure and ultimately died of cardiogenic shock. A focused autopsy was performed, revealing cardiomegaly, left ventricular hypertrophy (990 grams), right ventricular hypertrophy (10 mm) and increased ventricular septal thickness (maximum thickness 30 mm). There was extensive interstitial birefringent congophilic material in the left ventricle and interventricular septum, diagnostic of cardiac amyloidosis. The uninvolved myocardial fibers were hypertrophied with myocyte disarray, consistent with the diagnostic histological features of HCM (Figure 4 and Figure 5).
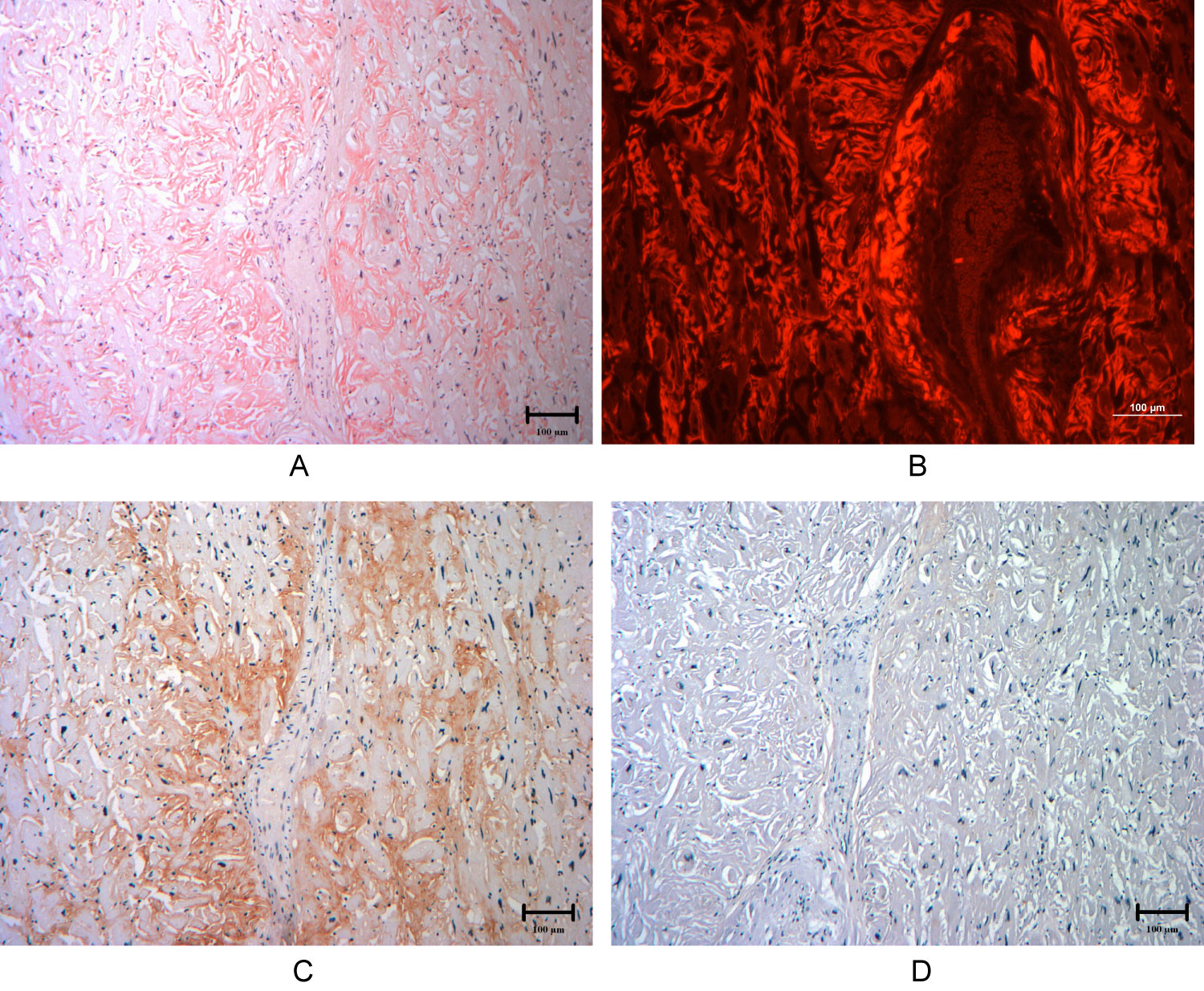 Figure 4: Cardiac histology samples from autopsy demonstrating extensive AL lambda amyloid deposition. Images are 100× magnification.
Figure 4: Cardiac histology samples from autopsy demonstrating extensive AL lambda amyloid deposition. Images are 100× magnification.
A) Extensive Congo red staining under white light, revealing extracellular amyloid fibril deposition; B) Congo red staining viewed under fluorescence light; C) Anti-lambda light chain antibody staining identifying lambda light chain fragments in the extracellular amyloid deposits, confirming the diagnosis of lambda AL cardiac amyloidosis; D) Anti-transthyretin light chain antibody reveals no significant transthyretin protein fragments, thus ruling out ATTR cardiac amyloidosis.
View Figure 4
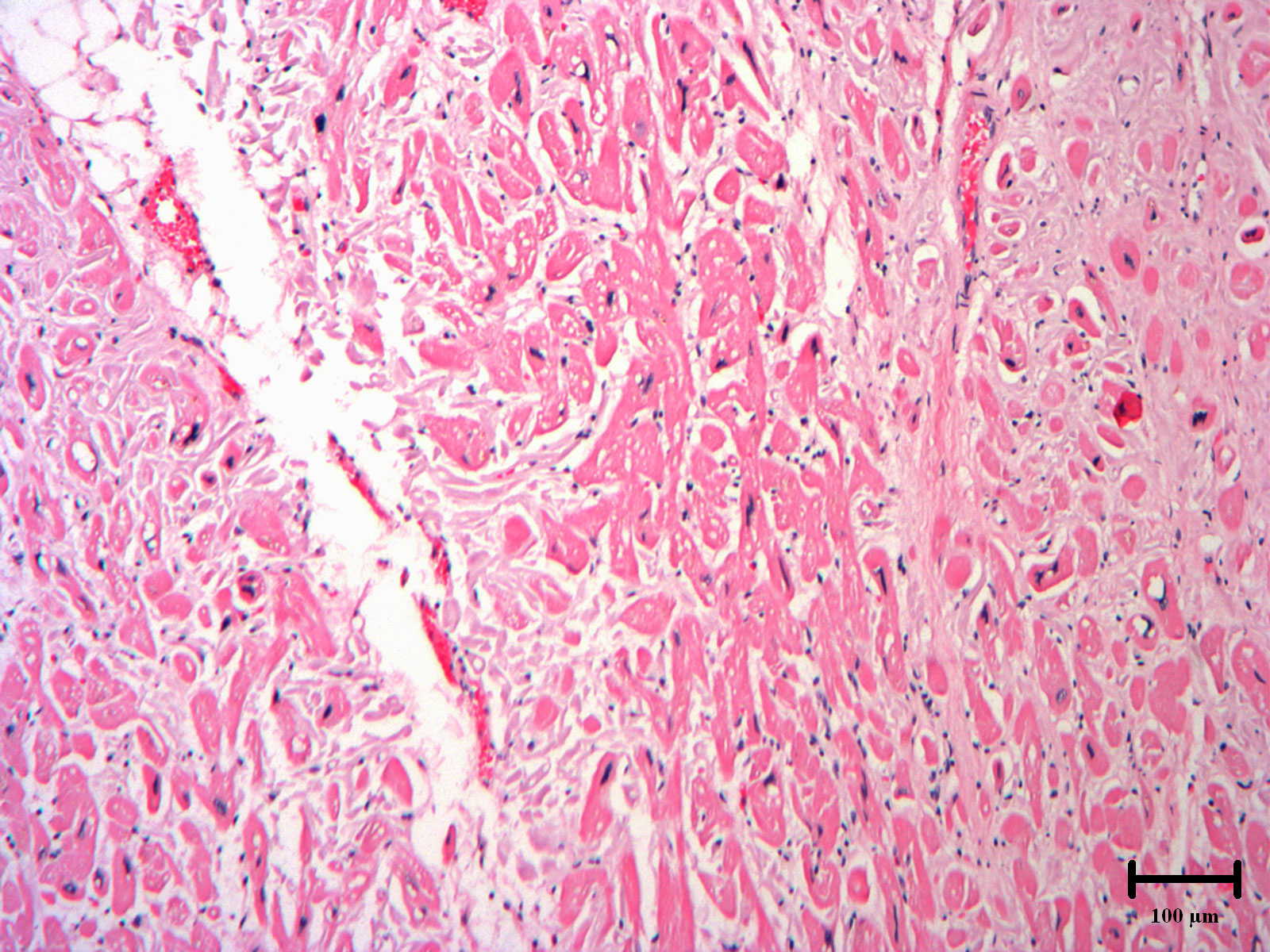 Figure 5: Autopsy heart section exhibiting the histology of hypertrophic cardiomyopathy. 100× magnification. H&E staining reveals enlarged and disarrayed myocytes with hyperchromatic nuclei and intercellular fibrosis.
View Figure 5
Figure 5: Autopsy heart section exhibiting the histology of hypertrophic cardiomyopathy. 100× magnification. H&E staining reveals enlarged and disarrayed myocytes with hyperchromatic nuclei and intercellular fibrosis.
View Figure 5
We present an unusual case of a woman with severely increased LV wall thickness and systemic AL amyloidosis who was ultimately diagnosed with both hypertrophic cardiomyopathy and AL cardiac amyloidosis at autopsy.
The systemic amyloidosis is a group of diseases caused by organ deposition of misfolded protein fragments. Disease manifestations and clinical course vary depending upon the precursor protein. In AL amyloidosis, the amyloidogenic protein is derived from monoclonal immunoglobulin light chains in the setting of a plasma cell dyscrasia. The incidence of AL amyloidosis is estimated to be about 9 cases per million people per year [2,3]. Tissue involvement with amyloid protein is diagnosed by light microscopy revealing characteristic apple-green birefringence with Congo red staining [4]. Cardiac involvement is common, affecting up to 50-70% of patients with systemic AL amyloidosis [5,6]. In contrast to AL amyloidosis, transthyretin (ATTR) cardiac amyloidosis is a distinct cause of cardiac amyloidosis which may be slowly progressive (particularly wild-type ATTR), and most resembles HCM in its echocardiographic appearance.
Cardiac amyloidosis is characterized by increased LV wall thickness, diastolic dysfunction, and low voltages on ECG [7]. The disease course in untreated patients is often one of rapidly progressive heart failure due to the underlying restrictive cardiomyopathy [8], often complicated by advanced conduction system disease and rarely, ventricular arrhythmias [9].
Hypertrophic cardiomyopathy (HCM) is a genetic cardiomyopathy caused by autosomal dominant mutations in genes encoding for the sarcomere and its associated myofibrils, with a prevalence of about 1 in 500 people worldwide [10]. While HCM is defined as LV hypertrophy (typically with septal wall thickness > 15 mm), in the absence of other cardiac or systemic diseases leading to LV hypertrophy, overlap between HCM and other causes of LV hypertrophy has been well described. A prior report from our institution demonstrates the phenotypic overlap that can occur between cardiac amyloidosis and HCM, with four out of 97 new cases of AL amyloidosis displaying echocardiographic findings typically associated with HCM, including LV outflow gradients and mitral systolic anterior motion [11]. There are few reports, of both HCM and amyloid cardiomyopathy occurring in the same patient [12], and even fewer with histopathological confirmation. In a case series of septal myectomy for presumed HCM, three out of 204 patients were found to have amyloid deposits, and all were over the age of 60-years with wild-type ATTR as the precursor protein [13].
On imaging, cardiac amyloidosis can be distinguished from HCM by traditional echo parameters, including more advanced LV diastolic dysfunction and low normal or impaired LV systolic function. There is considerable overlap in echocardiographic features, however, and these and other classic parameters lack sensitivity in distinguishing cardiac amyloidosis from HCM [14]. Recently, the inclusion of various cardiac deformation parameters increases the sensitivity and specificity for the detection of amyloidosis, especially the LVEF to global longitudinal strain ratio [14]. In addition, a non-invasive, diagnostic algorithm can accurately be used to diagnosis cardiac ATTR amyloidosis, even in cohorts with HCM [15].
Our patient had markedly increased ventricular wall thickness consistent with either cardiac amyloidosis or HCM. The patient had autopsy-proven, extensive extracellular amyloid deposition in the heart in the setting of known AL amyloidosis, consistent with a diagnosis of cardiac amyloidosis [16]. In addition to her cardiac amyloidosis, she was genotype and phenotype positive for HCM. The preponderance of clinical evidence suggested concomitant HCM as well: Asymmetric septal hypertrophy, systolic anterior motion of the mitral valve, and an autopsy revealing myocyte disarray characteristic of HCM in areas unaffected by amyloid deposition.
At initial presentation, this patient likely had HCM alone, given the significant interventricular septal thickness and minimal diastolic dysfunction. CMR supported this diagnosis, as there was only a single focus of delayed gadolinium enhancement within the anterior wall, as opposed to the diffuse delayed gadolinium enhancement that classically present in amyloid cardiomyopathy [16]. In patients with known systemic amyloidosis, CMR with late gadolinium enhancement has 86% sensitivity and 67% negative predictive value in diagnosis cardiac involvement [9]. The initial diagnosis of HCM was also supported by the fact that the patient maintained an excellent functional status for years, which was followed by the rapidly progressive clinical decline that characterizes AL cardiac amyloidosis. This clinical trajectory has also been described recently in another report of HCM and cardiac amyloidosis coexisting in the same patient [12].
This case highlights the importance of tissue diagnosis and genetic analysis in the evaluation of LV wall thickening when conflicting clinical features suggest divergent diagnoses. It also demonstrates the rapid clinical deterioration that can occur once a patient develops the superimposition of AL amyloid cardiomyopathy.
There was no support to disclosure: National Institutes of Health (NIH); Wellcome Trust; and other(s).
All authors precipitated in analysis and interpretation of the data, drafting the manuscript, acquiring key figures, and/or critical revisions for important intellectual content.