The inhibition of cyclooxygenases provides analgesic effect to relief inflammation and pain. The in silico study herein aimed to predict and elucidate the inhibitory potential of S. Sparganophora phytochemicals on the enzymes. An integrated molecular modeling approach which includes molecular docking, MMGBA, and pharmacokinetic profiling was employed to identify potential inhibitors of COX-1 and COX-2 from the characterized phytochemicals of S. Sparganophora. The results showed that myristic acid (-9.124), palmitic acid (-8.276), xanthinin (-8.063), 2,3-dehydro-4-oxo-beta-ionol (-7.596), coniferyl alcohol (-7.484) and alpha-D-glucopyranoside (-7.406) exhibited good binding to COX-1. Furthermore, alpha-D-glucopyranoside (-8.116), ambrosiol (-7.437), 2,3,5,5,8a-pentamethyl-6,7,8,8a-tetrahydro-5H-chromen-8-ol (-7.392), (7E)-2,6,6-trimethyl-3-methylene-7-(3-oxobutylidene) oxepanyl acetate (-7.392), (8S,14)-cedran-diol (-7.362), and 3-oxo-10(14)-epoxyguai-11(13)-en-6,12-olide (-7.183) ranked highly in binding to COX-2. It is worthy to note that alpha D-glucopyranoside showed robust binding to both protein targets and could be explored as a dual inhibitor of both proteins. Furthermore, pharmacokinetic studies showed that the reported compounds have good prospects of being oral anti-inflammatory drug candidates. These findings suggest that the reported compounds could be explored as inhibitors of COX-1 and COX-2.
Struchium sparganophora , Phytochemicals, Cyclooxygenases, Pharmacokinetic, Lipinski rule
Vegetables are the most important sources of vitamin A, a nutrient important for several metabolic activities in the body, in addition to its role as antioxidant; vegetables provide folate and potassium that are known to prevent birth defects, cancer, heart disease, hypertension and stroke; vegetables are good sources of phytochemicals [1-5]. A diet rich in vegetables and fruits can lower blood pressure, reduce the risk of heart disease and stroke, prevent some types of cancer, lower risk of eye and digestive problems, and have a positive effect upon blood sugar, which can help keep appetite in check. Most vegetables are naturally low in fat and calories [4,5]. Vegetables are important sources of many nutrients, including potassium, dietary fiber, folate, vitamin A, and vitamin C. Diets rich in potassium may help to maintain healthy blood pressure [5-9]. Struchium sparganophora , (Linn.) Asteraceae is a culinary herb consumed by Africans and it is known to have a nutritive and medicinal values. The green vegetable plays significant role in human nutrition, especially as a source of vitamins, minerals and dietary fibre. S . Sparganophora is a medicinal herb useful in the treatment of pain, fever, arthritis, rheumatism, neurological and mental disorders in traditional system of medicine in Nigeria and some other Africa countries [10,11]. Cyclooxygenase (COX) is an enzyme that helps create the chemicals prostaglandin and thromboxane (TxA2). Prostaglandins help create inflammation, and TxA2 helps in blood clot [12-15]. Cyclooxygenase (COX) enzymes, COX-1 and COX-2, are responsible for the catalysis of prostaglandin synthesis from its precursor arachidonic acid. COX-2 is normally undetectable in healthy tissue, though increased amounts of COX-2 can be detected in premalignant and in malignant tissue [12,16-18]. Both COX-1 and COX-2 produce the prostaglandins that contribute to pain, fever, and inflammation [12,15,19-21]. Cyclooxygenase metabolites have diverse effects in the lung and are known to modify airway tone, as well as inflammatory responses [15,16,22-25]. To the best of our knowledge, there is paucity information on the chemical composition and inhibitory potential of S. Sparganophora phytochemicals on cyclooxygenases so far. Therefore, this study was carried out to investigate and elucidate the inhibitory potential of S. Sparganophora phytochemicals on the enzymes (COX-1 and COX-2) to provide analgesic effect to relief inflammation and pain.
To identify potential inhibitors of COX-1 and COX-2, the compounds identified from our GC-MS analysis of S. Sparganophora were retrieved from Pub Chem repository in 2D sdf format [26,27]. Subsequently, the crystal structures of COX-1 (PDB ID: 1CQE) and COX-2 (PDB ID: 5KIR) were downloaded from Protein Data Bank ( http://www.rscb.org/ ).
The bioactive compounds of S. Sparganophora to be screened during the molecular docking were initially prepared using the functional Lig Prep tool in Maestro. In details, the ionization states and tautomers of the compounds were generated at pH = 7.2 ± 0.2 and subsequently optimized using the OPLS 2005 force field [27,28].
To prepare the targets for the molecular docking procedure, the protein structures were initially incorporated into Maestro and then prepared using the Protein Preparation Wizard. Specifically, the missing side chains were added using prime, solvents and other non-standard ligands were removed, hydrogen positions were optimized and restrained energy minimization was performed on the proteins [28]. Furthermore, grid boxes were generated using the position of the co-crystallized ligand of the proteins to map out the binding pockets and subsequently guide the automated docking procedure.
To identify compounds with potent inhibitory interactions with COX-1 and COX-2, the molecular docking procedure was carried out using the Glide script on maestro 11.1 [29]. The compounds were docked into the binding pockets of the protein targets guided by the prepared grid of the protein targets [30]. The results from the most rigorous screening (XP) were exported for further analysis.
The binding free energy change calculations were performed using Molecular Mechanics Generalized Born Surface Area (MM/GBSA) calculation [27,31-34]. The docked complexes were minimized by using local optimization feature in Prime wizard of Maestro [35]. The OPLS-2005 force field was employed to determine the binding energy for a set of receptor and ligand.
The binding free energy was estimated using the following equation:
∆G bind = ∆EMM + ∆GSolv + ∆GSA
Where, ΔEMM is the variance between the minimized energy of the protein-ligand complexes, while ΔGSolv is the variation between the GBSA solvation energy of the protein-ligand complexes and the sum of the solvation energies for the protein and ligand. In ΔGSA contains some of the surface area energies in the protein and ligand and the difference in the surface area energies for the complexes. The minimization of the docked complexes was done using a local optimization feature of prime [27,34].
The ADMET properties of the compounds were predicted using the user-friendly SWISSADME web tool ( http://www.swissadme.ch ) [36] and the Pro-Tox II web server ( https://tox-new.charite.de/protox_II/ ) [37]. In details, the properties predicted include lipophilicity, water solubility, gastrointestinal absorption, interaction CYP and P-glycoprotein, drug likeness and bioavailability score. Consensus log P, the arithmetic mean of different models of the partition coefficient of n-octanol to water was adopted as the measure of the lipophilicity of the compounds. ESOL model of water solubility (ESOL Log S) was used for solubility and Lipinski rule-based filter was adopted to predict the drug-likeness of the compounds. Furthermore, LD 50 , carcinogenicity, hepatotoxicity, mutagenicity, cytotoxicity, and immunotoxicity were predicted for the toxicity profiling.
The docking scores of the top-scoring compounds against both COX-1 and COX-2 are presented in Table 1. The reported compounds exhibit promising molecular binding to the proteins under study. However, for both protein targets, the standard co-crystallized ligand ranked higher than the test compounds in terms of binding affinity. In details, the standard ligand of COX-1 had a docking score of -12.212 which is relatively higher than the top scoring test compounds namely myristic acid, palmitic acid, Xanthinin,2,3-Dehydro-4-oxo-beta-ionol, Coniferyl alcohol, and Alpha-D-glucopyranoside which had docking scores of -9.124, -8.276, -8.063, -7.596, -7.484, and -7.406, respectively. In similar fashion, the co-crystalizedlig and of COX-2 had a docking score of -9.72 which is relatively higher than alpha-D-glucopyranoside, ambrosiol, 2,3,5,5,8 a-pentamethyl-6,7,8,8a-tetrahydro-5H-chromen-8-ol, (7E)-2,6,6-Trimethyl-3-methylene-7-(3-oxobutylidene) oxepanyl acetate, (8S,14)-cedran-diol, and 3-oxo-10(14)-epoxyguai-11(13)-en-6,12-olide with docking scores of -8.116, -7.437, -7.392, -7.382, -7.352 and -7.173 respectively. Interestingly, the binding affinities of our test compounds to both COX-1 and COX-2 ranked higher than the affinity of the test compounds reported by Omoboyowa on the same targets [38]. The 2D amino acid interaction diagrams (Figure 1 and Figure 2) showed that hydrogen bond is the most prevalent interaction observed in all interactions. It is, however, worthy to note that Alpha D-glucopyranoside exhibited impressive binding to both COX-1 and COX-2 and could be explored as a dual inhibitor of both proteins. The generated poses of the ligands bound to the binding site of the protein targets were re-scored using MMGBSA calculations. This protocol is not new and has shown promising results in identifying small molecule inhibitors of a wide range of proteins. The prime MMGBSA analysis showed the relative binding-free energy (ΔG bind) of each ligand in complex with the ligand-binding site of both target proteins (COX-1 and COX-2), and the results are presented in Figure 3. The trend in the results is similar to that which was obtained from the molecular docking. The standard co-crystallized ligand exhibited the highest binding of all compounds tested against COX-1 with a ΔG value of -55.733 kcal/mol. Myristic acid, palmitic acid, xanthinin, 2,3-Dehydro-4-oxo-beta-ionol, coniferyl alcohol, and alpha-D-glucopyranoside had energy values of -29.940 kcal/mol, -14.106 kcal/mol, -12.934 kcal/mol, -38.024 kcal/mol, -48.711 kcal/mol and -31.340 kcal/mol, respectively. Coniferyl alcohol had the closest binding energy to the co-crystalized ligand. In similar fashion, the co-crystallized ligand of COX-2 also had the highest binding to COX-2 with a ΔG value of -63.290 kcal/mol. Ambrosiol and (8S,14)-cedran-diol showed “positive” ΔG values of 9.282 kcal/mol and 10.712 kcal/mol respectively. 2,3,5,5,8a-pentamethyl-6,7,8,8a-tetrahydro-5H-chromen-8-ol was found to have a similar binding energy to (7E)-2,6,6-Trimethyl-3-methylene-7-(3-oxobutylidene) oxepanyl acetate with a ΔG value of -37.675 kcal/mol. This was observed to be the closest binding energy to the standard co-crystallized ligand. The amino acid interactions of the lead compounds with the ligand binding site of COX-1 and COX-2 were analyzed to identify optimizable ligand-protein interactions. The identification and optimization of the specific interactions can improve the binding affinity of the compounds to their bound receptors. Palmitic acid had two hydrogen bond interactions with the binding pocket of COX-1 (Figure 1) with both bonds occurring as a result of donations from ARG 120 and TYR 355 to the carboxylic group of the compound. Myristic exhibited exactly the same binding conformation as Palmitic acid. Coniferyl alcohol had one hydrogen bond interaction with TYR 355. Similarly, Xanthinin showed only one hydrogen bond interaction with SER 530. Furthermore, alpha-D-glucopyranoside had interactions with three amino acids; a hydrogen bond donation to MET 522, an accepted hydrogen bond from ARG 120 and a pi-stacking between the aromatic ring of the compound and TYR 355, an aromatic amino acid residue. 2,3-Dehydro-4-oxo-beta-ionol had just one interaction with the binding pocket of COX-1; a hydrogen bond accepted from SER 530. Remarkably, these amino acid interactions have been reported in the inhibition of COX-1 [38]. In its characteristic binding to the ligand-binding site of COX-2, alpha-D-glucopyranoside, the proposed dual inhibitor made contact with three amino acid residues; hydrogen bond donations to SER 353 and GLN 192 and a pi-stacking between the aromatic group of the compound and TRP 387, an aromatic amino acid residue. Also, (8S,14)-cedran-diol exhibited one hydrogen bond interaction with MET 522. However, no hydrogen bond interaction was observed in the binding of ambrosiol, 2,3,5,5,8a-pentamethyl-6,7,8,8a-tetrahydro-5H-chromen-8-ol, (7E)-2,6,6-trimethyl-3-methylene-7-(3-oxobutylidene) oxepanyl acetate, and 3-oxo-10(14)-epoxyguai-11(13)-en-6,12-olide to the pocket of COX-2. More rigorous molecular modeling approach might be required to fully understand and elucidate the specific binding characteristics of these compounds.
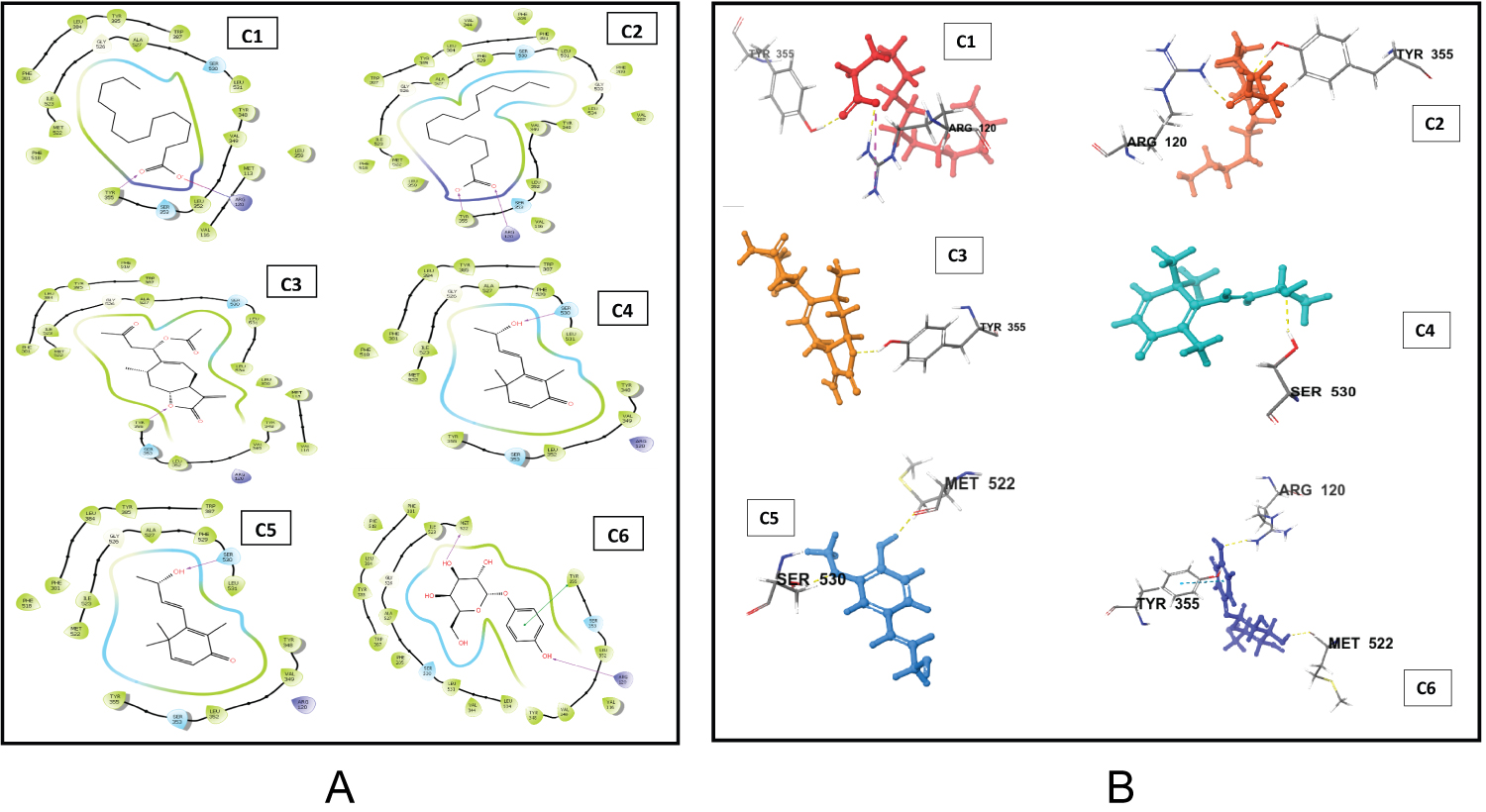 Figure 1: (A) 2D interactions of the the lead compounds with the ligand binding site of COX-1; (B) 3D interactions of the lead compounds with the ligand-binding site of COX-1.
Figure 1: (A) 2D interactions of the the lead compounds with the ligand binding site of COX-1; (B) 3D interactions of the lead compounds with the ligand-binding site of COX-1.
C1 = Myristic Acid, C2 = Palmitic Acid, C3 = Xanthinin, C4 = 2,3-Dehydro-4-oxo-beta-ionol, C5 = Coniferyl alcohol, C6 = Alpha-D-glucopyranoside
View Figure 1
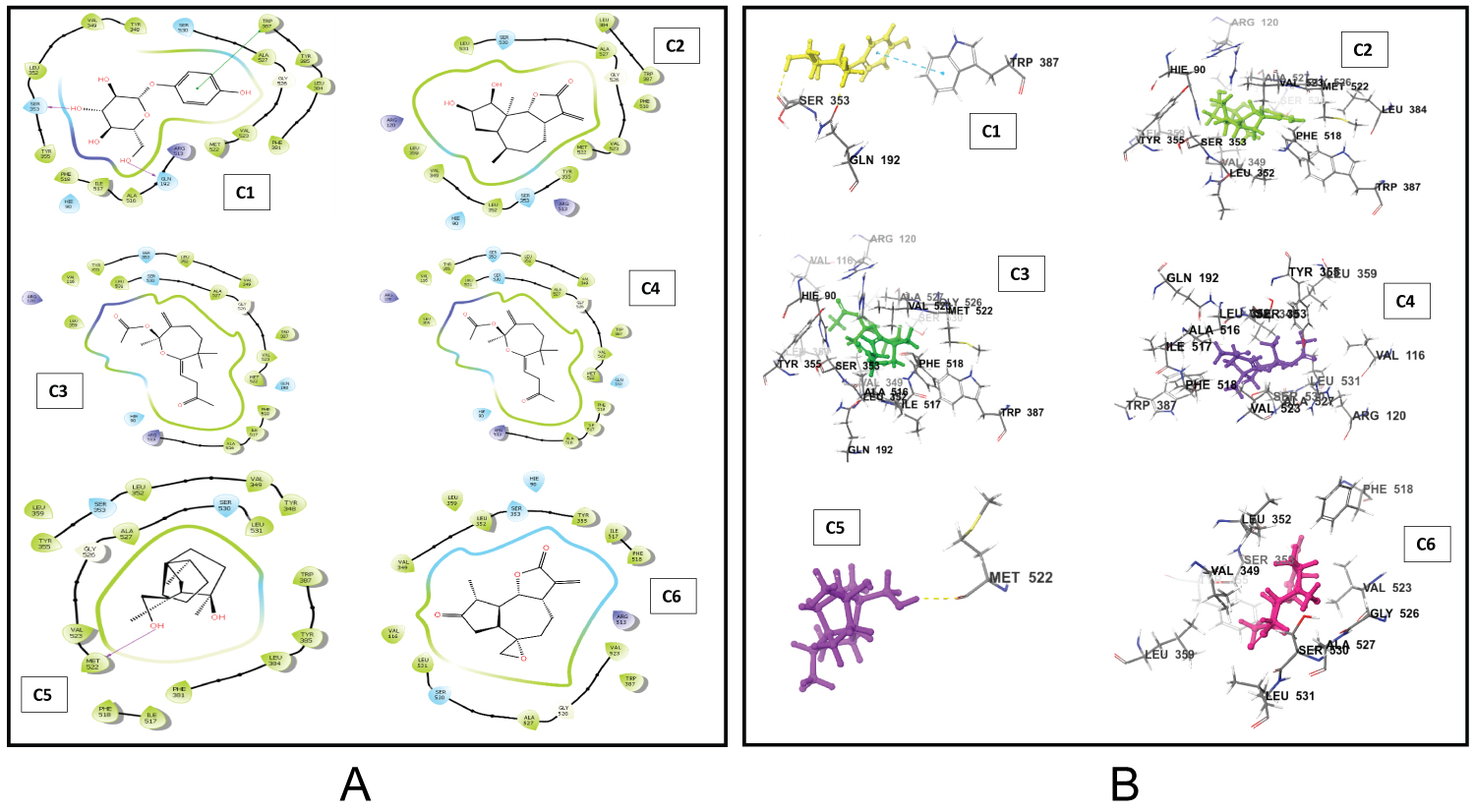 Figure 2: (A) 2D interactions of the lead compounds with the ligand binding site of COX-2; (B) 3D interactions of the lead compounds with the ligand binding site of COX-2.
Figure 2: (A) 2D interactions of the lead compounds with the ligand binding site of COX-2; (B) 3D interactions of the lead compounds with the ligand binding site of COX-2.
C1 = Alpha-D-glucopyranoside, C2 = Ambrosiol, C3 = 2,3,5,5,8a-pentamethyl-6,7,8,8a-tetrahydro-5H-chromen-8-ol, C4 = (7E)-2,6,6-Trimethyl-3-methylene-7-(3-oxobutylidene)oxepanyl acetate, C5 = (8S,14)-cedran-diol, C6 = 3-oxo-10(14)-epoxyguai-11(13)-en-6,12-olide.
View Figure 2
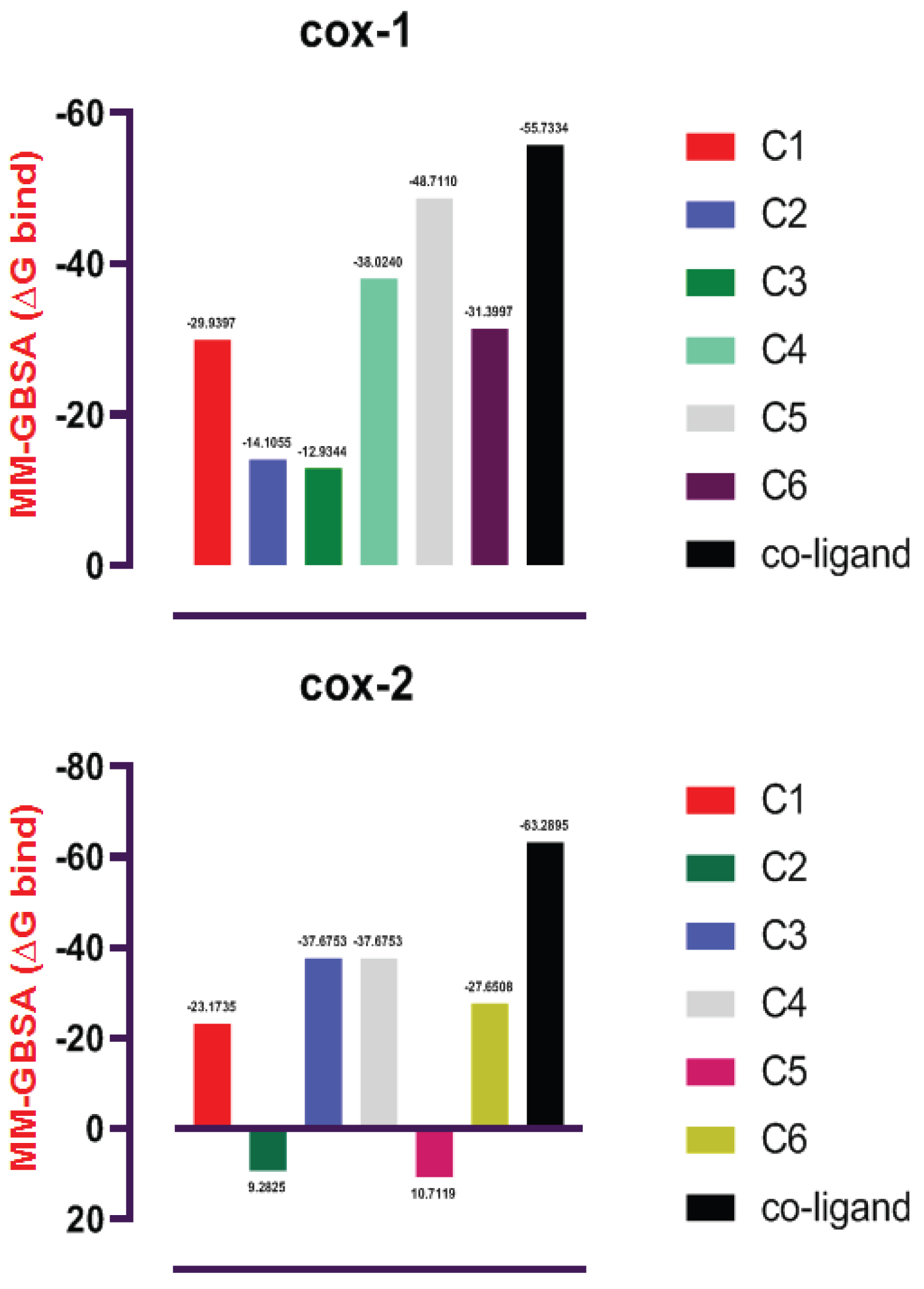 Figure 3: COX-1: C1 = myristic acid, C2 = palmitic acid, C3 = Xanthinin, C4 = 2,3-Dehydro-4-oxo-beta-ionol, C5 = Coniferyl alcohol, C6 = alpha-D-glucopyranoside
Figure 3: COX-1: C1 = myristic acid, C2 = palmitic acid, C3 = Xanthinin, C4 = 2,3-Dehydro-4-oxo-beta-ionol, C5 = Coniferyl alcohol, C6 = alpha-D-glucopyranoside
COX-2: C1 = Alpha-D-glucopyranoside, C2 = Ambrosiol, C3 = 2,3,5,5,8a-pentamethyl-6,7,8,8a-tetrahydro-5H-chromen-8-ol, C4 = (7E)-2,6,6-Trimethyl-3-methylene-7-(3-oxobutylidene)oxepanyl acetate, C5 = (8S,14)-cedran-diol, C6 = 3-oxo-10(14)-epoxyguai-11(13)-en-6,12-olide.
View Figure 3
Table 1: Docking scores of the test compounds. View Table 1
The lipophilicity and water solubility of the lead compounds are presented in Table 2. All the compounds showed good levels of lipophilicity required to have a smooth movement across the intestinal barrier. Palmitic acid was predicted to be the most lipophilic with a log P value of 5.2 and alpha-D-glucopyranoside the least lipophilic with a value of -0.17. A drug candidate must have a sufficient lipophilicity that aids its transport from the gastrointestinal tract into systemic circulation. Complementarily, water solubility facilitates the movement of the drug molecule in the hydrophilic condition of the blood. Drugs are transported to their sites of action through the blood by binding to resident transport molecules in the blood such as albumin. As per ESOL model of water solubility, alpha-D-glucopyranoside showed the highest level of water solubility with a value of -0.71 and palmitic, the least soluble with an ESOL Log S value of -5.02.
Table 2: The physicochemical properties of the lead compounds. View Table 2
The pharmacokinetic profiles of the top-scoring compounds are presented in Table 3. All compounds have a high gastrointestinal absorption. Oral drug candidates must have a structural orientation which enables them cross the lumen of the small intestine into systemic circulation. A high gastrointestinal absorption results from a good lipophilicity-hydrophilicity ratio. Also, none of the compounds is predicted to be a substrate of permeability glycoprotein. This protein is a member of the ATP-binding cassette proteins which orchestrates the active transport of compounds out of the cell before reaching a significant therapeutic concentration in the cell. Xanthinin, 2,3-Dehydro-4-oxo-beta-ionol, Coniferyl alcohol, alpha-D-glucopyranoside, Ambrosiol, (8S,14)-cedran-diol, and 3-oxo-10(14)-epoxyguai-11(13)-en-6,12-olide are non-inhibitors of the vital CYP isoforms. This family of enzymes catalyze phase 1 reaction of drug metabolism which contributes substantially to the clearance of the compounds. Inhibiting any of the participating isoforms can induce a drug-drug interaction which elicits the toxic effects of the drug due to its delayed clearance.
Table 3: The pharmacokinetic profiles of the top-scoring compounds. View Table 3
The drug likeness and bioavailability score of the top-scoring compounds are presented in Table 4. These complementary properties predict the oral drug candidacy of small molecular weight compounds. The result showed that palmitic acid violated one of the Lipinski rule-of-five where all the other compounds violated none of the rules. The Lipinski rule of five for drug-likeness showed that all the compounds are drug-like. The rule is valid when mol. MW < 500, QPlogPo/w < 5, donor HB ≤ 5, acceptor HB ≤ 10 [39]. This rule-based filter is constantly employed in drug design to predict the likelihood of a compound being an oral drug candidate. Furthermore, all the reported compounds had positive bioavailability scores. Myristic acid and palmitic acid had a bioavailability score of 0.85 while the other compounds returned a bioavailability score of 0.55. In general, a bioavailability score of at least 0.10 is required to be considered a candidate [40].
Table 4: The druglikeness and bioavailability score of the test compounds. View Table 4
The toxicity profiles of the compounds are presented in Table 5. Generally, Pro Tox-II presents the mutageni city, carcinogenicity, immunotoxicity, hepatotoxicity, and cytoxicity profiles of chemical compounds. Additionally, it places chemical compounds into 6 classes of toxicity, with Class 1 being the most toxic and Class 6 being the least toxic. Interestingly, all the lead compounds are generally less toxic with LD 50 (mg/kg) ranging from 900 to 23,000 and toxicity class between 4 to 6, except C7 and C11 with LD 50 of 150 mg/kg and toxicity class 3. However, none of the compounds is found to be carcinogenic, hepatotoxic, mutagenic or cytotoxic, though C3, C5, C7, and C11 are predicted to be immunotoxic.
Table 5: The toxicity profiles of the lead compounds. View Table 5
The investigated phytochemicals in S . Sparganophora exhibit promising pharmacological potential to inhibit COX-1 and COX-2. The binding affinities of our test compounds to both COX-1 and COX-2 ranked higher. All compounds have a high gastrointestinal absorption. The result showed that almost all the compounds violated none of the Lipinski rules. None of the compounds is found to be carcinogenic, hepatotoxic, mutagenic or cytotoxic. Pharmacokinetic studies showed that the reported compounds have good prospects of being oral anti-inflammatory drug candidates. These findings suggest that the reported compounds could be explored as inhibitors of COX-1 and COX-2.
We have no conflict of interest.
The authors confirm that the data supporting the findings of this study are available within the article.