The neurobiological features of Post-Traumatic Stress Disorder (PTSD), a debilitating psychiatric condition triggered by exposure to traumatic events, are associated with enigmatic pathological mechanisms which are becoming clearer as the library of associated literature increases. This article considers the clinical features of PTSD, the neural circuitry at work, and some of the genetic and epigenetic factors contributing to PTSD risk. In addition, this review presents various models proposed to explain the progression of PTSD, including the Synaptic Model of Chronic Stress Pathology, the Network-based Model, and the Dual Pathology Model.
This review underscores the need for more objective evidence of risk stratification and diagnosis by elucidating recent research into sleep disturbances and potential biomarkers associated with PTSD. The present review concludes that the neurobiological and genetic mechanisms which underlie PTSD are crucial to aid in developing novel, effective treatments and risk stratification tools, and that biomarker analysis holds promise for enhancing the identification of individuals at risk and providing targeted interventions.
Post-traumatic stress disorder (PTSD), Biomarkers, Sleep disturbances
Post-Traumatic Stress Disorder (PTSD) is a debilitating, psychiatric condition triggered by exposure to, observing or learning about a traumatic event e.g., active combat, sexual violence, road traffic collisions or violent assaults [1,2].
The symptoms associated with PTSD vary widely depending on the severity and the trauma type to which an individual has been exposed. However, commonly observed symptoms include sleep disturbances, nightmares, problems falling and staying asleep, flashbacks, severe anxiety, panic attacks, and intrusive memories relating to the event [3]. In many potential trauma events (PTE), symptoms self-resolve within one month of the traumatic event. These cases are known as acute stress disorder and are considered independent of PTSD [4]. Symptoms persisting ≥ 1 month are essential for a diagnosis of PTSD [4].
Almost 70% of the global population has been or will be, exposed to a PTE and almost 6% of those will go on to develop PTSD [3]. However, these estimates seem unrealistic as they do not consider demographics. PTSD prevalence in physical assault victims, combat veterans and rape victims are estimated to be 20%, 25% and 50% respectively [3]. Data would suggest that PTSD risk is more closely associated with the type and severity of trauma rather than an individual’s propensity for the disorder. Nonetheless, studies have identified several risk factors relating to PTSD [1,3,4].
It is estimated that up to 40% of PTSD risk is genetic [4]. Several genetic and epigenetic alterations have been shown to be associated with increased or decreased risk of developing PTSD, as well as physiological factors including low heart rate variability and low cortisol levels. In addition, PTSD prevalence in women versus men is estimated at around 2:1 [3]. The risk of developing this multi-factorial condition is also dependent on the severity and nature of the trauma, history of childhood trauma, low social support, and a family history of trauma [5].
The neural circuitry relating to PTSD is becoming clearer as our understanding of the brain and its processes are explicated. Helpfully, PTSD is largely associated with the fear circuitry which is highly conserved in mammals and is one of the most well-understood mechanisms of brain function [4].
Diagnosis of PTSD takes the form of the ‘CAPS-5 case report form - PTSD screening’ in which a series of questions are posed to the individual and a score is provided. This score can then be used to stratify the risk to the individual and confirm or reject a PTSD diagnosis. This method is subject to interpretation by the clinician and to the disposition of the individual in question. Therefore, there is a need for a more definitive method of risk stratification and diagnosis.
This article discusses the major neurobiological regions and mechanisms associated with PTSD. In addition, this article investigates various proposed models of PTSD and elucidates recent research in which biomarkers relating to PTSD risk have been identified.
The clinical features of PTSD can be categorised as shown in Table 1.
Table 1: Clinical features of PTSD. View Table 1
Research has revealed that these features are the result of several interconnected phenomena including difficulties in fear extinction, where the ability to diminish fear responses is impaired [1,3,4]. Additionally, there is an increased tendency to generalise fear responses to stimuli that are not directly associated with the traumatic event. Furthermore, people with PTSD tend to exhibit a negative bias, perceiving neutral stimuli as threatening and experiencing a sense of danger even in safe environments [4].
There are various neural mechanisms proposed to explain the development of PTSD. Several parts of the brain including the amygdala, hippocampus, hypothalamus, and prefrontal cortex are involved in these neural mechanisms (Figure 1).
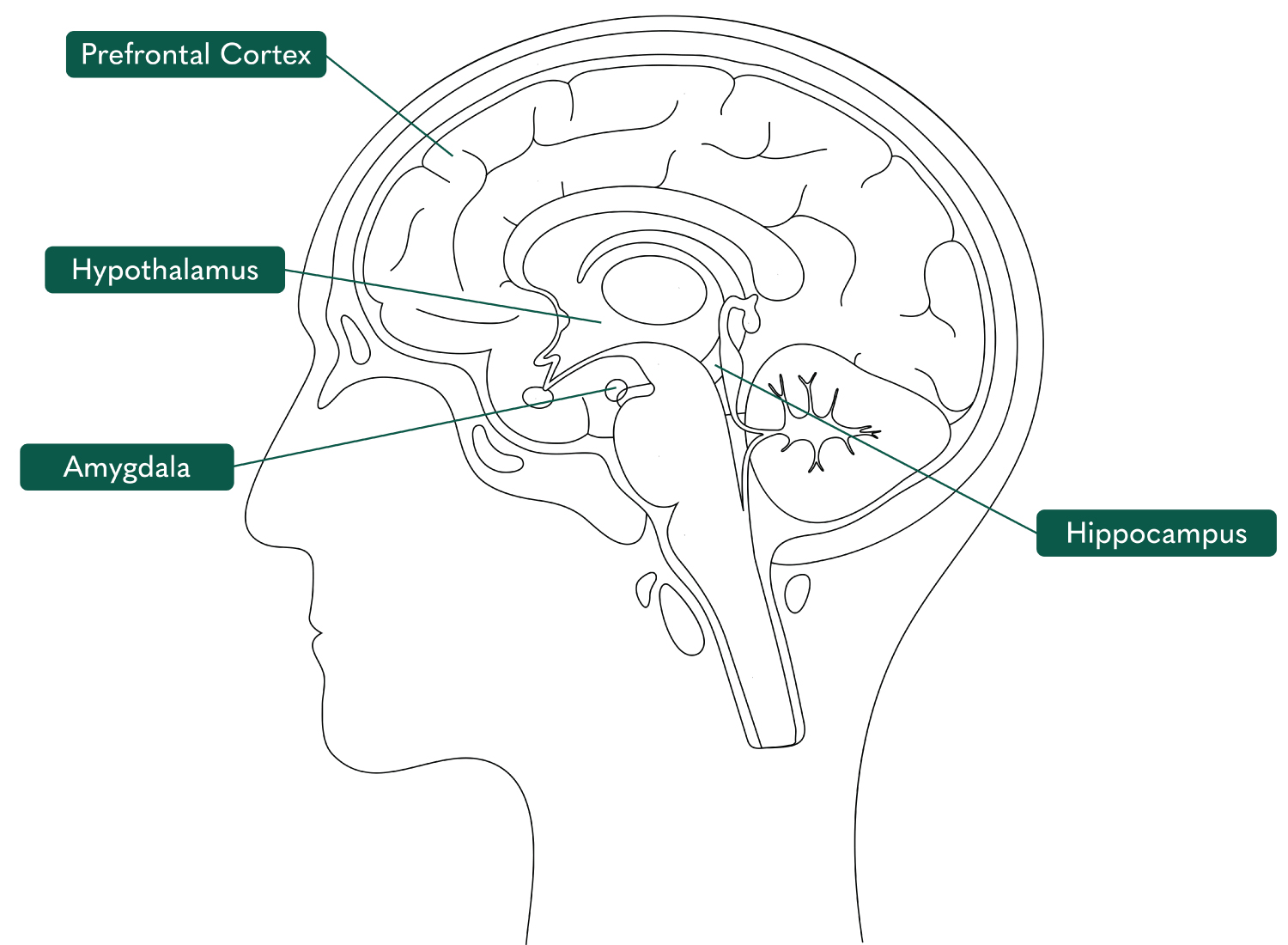 Figure 1: Illustration of the brain marking the prefrontal cortex, hypothalamus, amygdala, and hippocampus [6].
View Figure 1
Figure 1: Illustration of the brain marking the prefrontal cortex, hypothalamus, amygdala, and hippocampus [6].
View Figure 1
The amygdala is regarded as the threat-processing centre of the brain, therefore its connection with PTSD is significant. Conditioned and unconditioned stimuli converge in the lateral nucleus of the amygdala which subsequently transmits to the central amygdala before sending output signals to the hypothalamus, brainstem, and other crucial areas of the brain [7]. Amygdala hyperactivity has been reported in PTSD patients when compared with non-exposed individuals and with those who have experienced trauma but have not developed PTSD [4]. However, it is unclear whether this is causal or a consequence of PTSD. Many of the physiological responses associated with PTSD are the result of downstream pathways of the amygdala. These pathways lead to many of the fear reflexes associated with PTSD including elevated heart rate by projections to the hypothalamus, locus coeruleus and dorsal vagal nerve, increased startle response through projections to the reticularis pontis caudalis, elevated breathing rate through parabrachial connections and activation of the hypothalamic-pituitary-adrenal axis through projections to the paraventricular nucleus of the hypothalamus [4].
The hypothalamic-pituitary-adrenal (HPA) axis is a complex collection of endocrine signals and feedback loops incorporating three components, the hypothalamus, the pituitary gland and the adrenal gland, that regulate the body’s response to stress. In healthy individuals, the normal stress response proceeds as follows (Figure 2). Once triggered by a stressor, the hypothalamus increases the expression of corticotropin-releasing hormone (CRH) through inputs from the amygdala. CRH then activates the anterior pituitary gland, increasing the production of adrenocorticotropic hormone (ACTH) which subsequently signals the adrenal medulla, liberating cortisol expression. Cortisol then signals the hypothalamus, exerting a negative feedback loop and completing the self-regulating process [8]. Those at increased vulnerability to PTSD display significantly lower cortisol expression and dysregulation of the processes involved in this stress response [1]. Additionally, alterations in CRH expressions may promote dysregulation of the startle response, hyperarousal, and anxiety; the central features of PTSD [9].
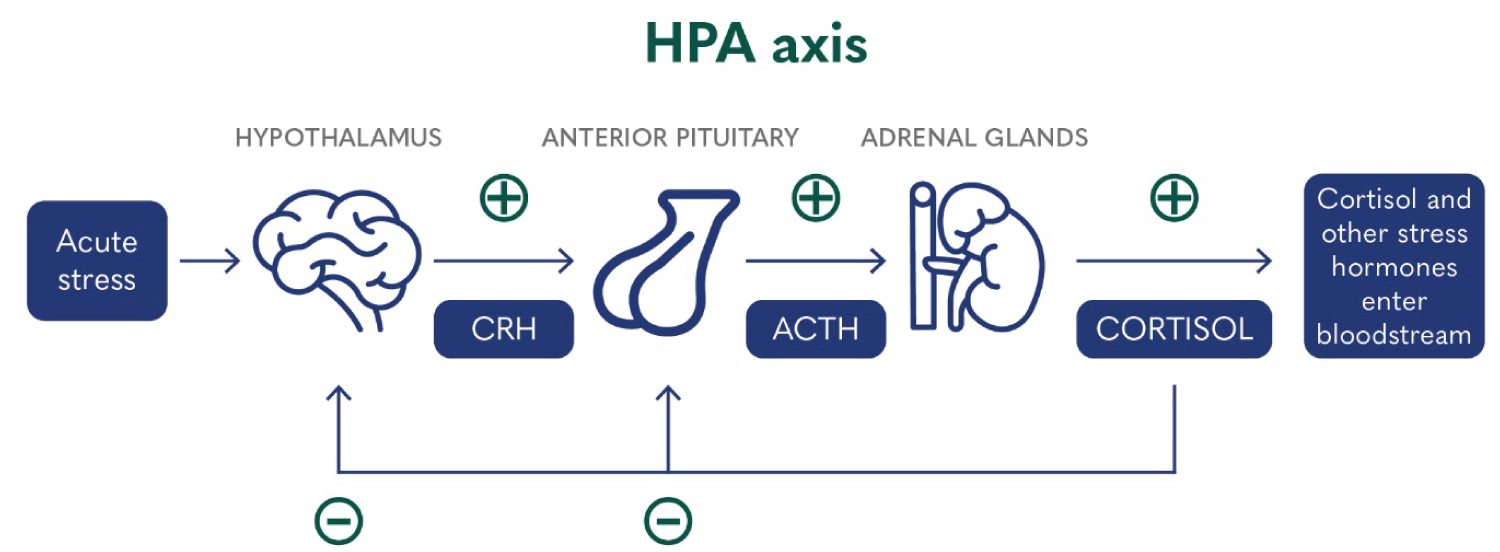 Figure 2: Illustration of the HPA axis and its function in the normal stress response [10].
View Figure 2
Figure 2: Illustration of the HPA axis and its function in the normal stress response [10].
View Figure 2
Several parts of the prefrontal region of the brain have been associated with PTSD. The anterior cingulate cortex (ACC) displays inhibitory control over the stress response and emotional reactivity through regulation of the amygdala, allowing for fear responses to be diminished or eliminated. The decreased ACC volume observed in PTSD patients is thought to interrupt these processes, contributing to PTSD pathogenesis. The ACC is also responsible for the active learning process which is impaired in PTSD [1].
The human medial prefrontal cortex (MPC), also located in the prefrontal region, is crucial for fear processing. Working in tandem with the hippocampus, the MPC provides an inhibitory mechanism of control relating to threat-related memories and behaviours [4]. Decreased activation of the MPC, in particular the subgenual prefrontal cortex, has been shown to be underactive in PTSD patients [4]. The MPC is also involved in the regulation of emotion and arousal and has, therefore, been implicated in the emotional overmodulation displayed by dissociative forms of PTSD.
Since neuroimaging studies of PTSD patients began, the hippocampus has been identified as being important in PTSD pathophysiology. The hippocampus is associated with long-term memory formation and memory processing [1]. Smaller hippocampal volumes have been identified in PTSD patients and numerous clinical studies have reported a strong correlation between trauma and lower hippocampal volumes [4,11,12]. This pre-existing hippocampal deficit may be responsible for the impaired contextualisation and interpretation of one’s trauma [13]. Furthermore, PTSD patients have been shown to display unusual brain activity in the hippocampal and parahippocampal regions of the brain, suggesting an effect on the formation of traumatic memories [1].
Catecholamines are monoamine neurotransmitters responsible for conveying signals from neurons to other cells throughout the body. They are crucial in the normal stress response, actioning physiological responses such as increased heart rate and skin conductance.
Norepinephrine (Figure 3) is the principal driver of the autonomic stress response in the central nervous system and periphery. Norepinephrine neurons project from the locus coeruleus to the prefrontal cortex (PFC), amygdala, hippocampus and hypothalamus and norepinephrine is responsible for the encoding of memories and promotion of hypervigilance. In PTSD patients, levels of norpepinephrine are elevated, facilitating a deeper encoding of the traumatic memories. Peripheral norepinephrine, produced by the adrenal medulla, is responsible for increased heart rate, blood pressure and skin conductance, features frequently associated with PTSD [14].
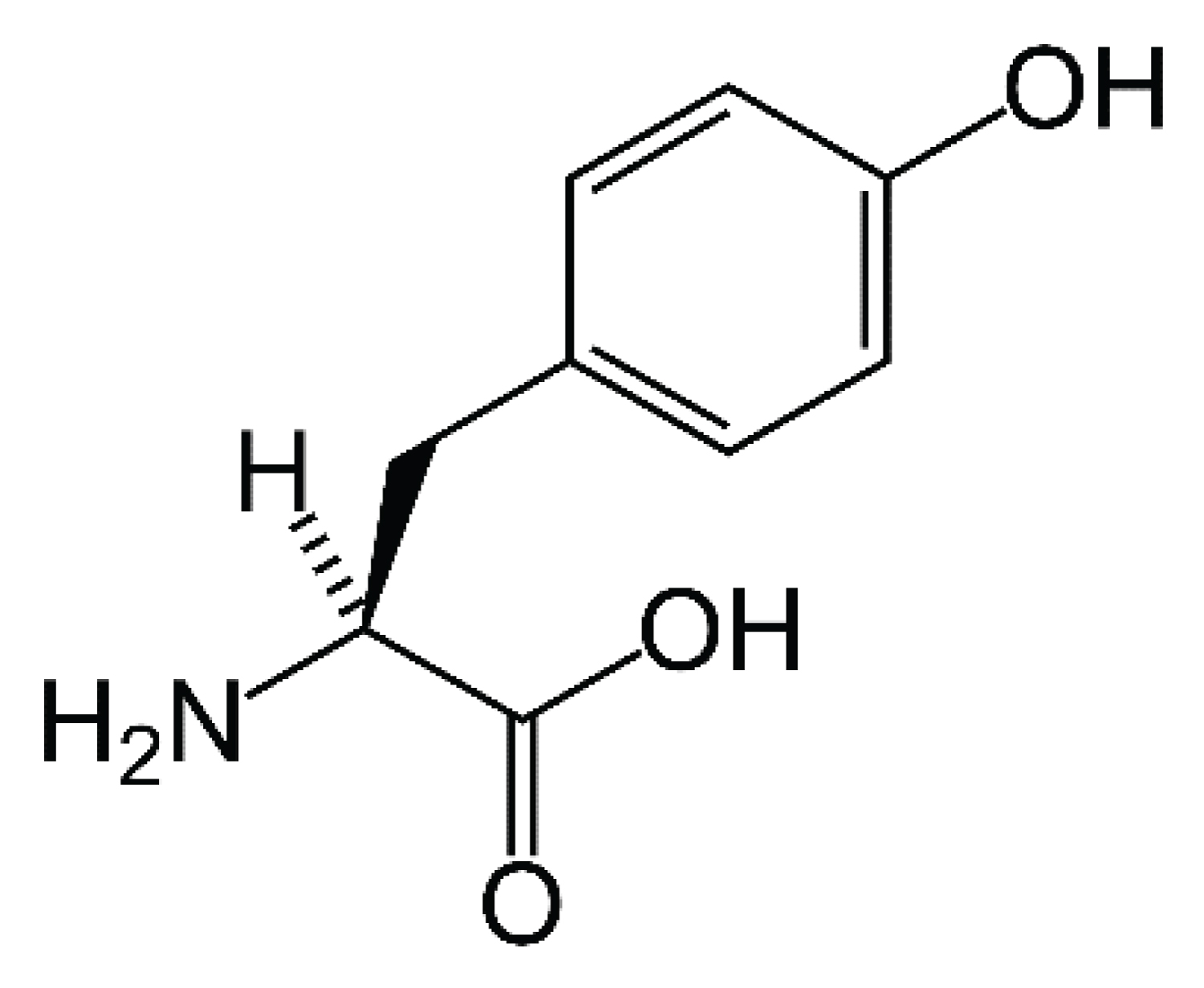 Figure 3: Chemical structure of norepinephrine.
View Figure 3
Figure 3: Chemical structure of norepinephrine.
View Figure 3
Other neurotransmitters implicated in PTSD include gamma-aminobutyric acids (GABA) receptors, glutamate and neuropeptide Y. Lower concentrations of GABA receptors are reported in PTSD patients when compared with healthy controls [15]. Glutamate dysregulation is increasingly being considered as paramount to the development of PTSD [3] and neuropeptide Y circulatory levels have been shown to be decreased in PTSD patients [16].
It is also of interest to note that endogenous opioid levels may play a protective role in the development of PTSD. One study reported that acute morphine administration had protective effects on PTSD, limiting fear conditioning in the aftermath of a traumatic injury, suggesting a protective effect on severity, but not development [17]. Another investigation, which studied veterans of the war in Iraq, suggested that the use of morphine during trauma care may alleviate the risk of the development of PTSD post-traumatic injury [18]. While this is not sufficient to be considered evidence for the protective effect of opioids, it certainly warrants further investigation.
A significant proportion of PTSD risk is attributed to genetics and epigenetic alterations. Firstly, gene variants of glucocorticoid receptors have been implicated in PTSD risk. Variations in the FKBP5 gene were originally associated with PTSD patients who experienced child abuse but have since been shown to be associated with several aspects of PTSD development, for example, the type and severity of PTSD symptoms, startle response and neural activity [4].
Variants of the COMT gene, namely COMT Val 158 Met polymorphism, have been linked to an increased risk of developing PTSD and a low expression variant in the serotonin transporter gene has been linked with an increased risk of depression and increased amygdala activity [19].
Epigenetic changes are also associated with PTSD. DNA methylation typically functions to silence the expression of genes. One study looked at DNA methylation in the offspring of individuals who suffer from PTSD. This study reveals that parental PTSD can lead to differential DNA methylation of the glucocorticoid receptor gene in offspring, indicating that PTSD-related epigenetic changes can be inherited. This underscores the biological complexity of PTSD, extending beyond a mere psychological condition to involve significant genetic alterations [20].
The pathology of PTSD is complicated. Evidence strongly implicates stress in the pathophysiology of PTSD with glutamate dysregulation and synaptic loss playing an important role. Herein, we discuss some of the established models of PTSD.
Neuronal remodelling can be induced by extreme and repeated stressors, resulting in increased synaptic density. Prolonged stress responses have been linked to various effects within the PFC and hippocampus. These include disturbances in glucocorticoid signalling, heightened neuroinflammation, decreased levels of brain-derived neurotrophic factor (BDNF), deficits in astrocytic function, and diminished uptake of synaptically-released glutamate. Consequently, these changes can result in elevated extracellular glutamate levels and the potential for excitotoxicity, which refers to excessive activation of neurons leading to cellular damage [21]. During a prolonged stress response, an intriguing phenomenon occurs where extracellular glutamate levels paradoxically rise, despite a substantial decrease in glutamate neurotransmission, as well as diminished activity of N-methyl-D-aspartate receptors and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. These molecular alterations lead to neuronal atrophy, which is characterized by a decrease in dendritic length and arborization, and a reduction in synaptic density and neurotransmission strength. Consequently, this loss of synapses and hypoconnectivity directly correlates with behavioural abnormalities, specifically mood and anxiety dysregulation [3].
CSP in the amygdala manifests as functional and structural alterations. Specifically, there is a notable decrease in synaptic connectivity within the medial amygdala, while the basolateral amygdala shows increased levels of brain-derived neurotrophic factor and enhanced synaptic connectivity [3].
A solitary stressor is adequate to trigger an upregulation of BDNF in the basolateral amygdala, observable within one day and persisting for a duration of ten days. Furthermore, this single stressor progressively promotes synaptogenesis in the basolateral amygdala over the course of ten days, coinciding with a gradual increase in anxiety-like behaviour [3].
Furthermore, following the cessation of the stressor, the synaptic loss in the hippocampus and prefrontal cortex, the downregulation of BDNF, and the associated behavioural disturbances show signs of recovery within a span of two to four weeks. However, in contrast, the synaptic hyperconnectivity and anxiety-related behaviour observed in the basolateral amygdala are not reversible within the same timeframe [3].
Studies show that trauma-related synaptic dysconnectivity, both in terms of structure and function, occur in animals and humans within brain regions that are essential for regulating anxiety, mood, and fear. It is thought that decreased synaptic connectivity in the hippocampus, coupled with increased synaptic connectivity in the amygdala, could act as predisposing factors. These factors may be further intensified by the occurrence of a traumatic event, ultimately leading to the disruption of fear memory function [3].
In those with PTSD, heightened fear and arousal are believed to be associated with increased activity in the amygdala and dorsal anterior cingulate cortex. This hyperactivity is thought to be a result of diminished regulation from a less active prefrontal cortex, as well as deficits in fear extinction driven by dysfunction in the medial PFC and hippocampus [3].
A comprehensive study of the PTSD pathophysiology literature has resulted in a number of apparently contradictory findings. In many cases, reduced hippocampal volume [11,12] is seen as a driving factor for the progression of PTSD, however, some studies show no significant change [22]. Similarly, some studies report increased amygdala volume [23] in their findings, whilst others report decreased volumes [11,24,25].
This contradiction prompted the proposal of the following ‘Dual Pathology Model’ by Abdallah, et al. (2019). This model highlights the association between synaptic changes and two distinct types of pathology: amino acid-based pathology (ABP) and monoamine-based pathology (MBP).
ABP is characterized by glutamate dysregulation and synaptic loss, which is evidenced by treatment resistance to monoaminergic drugs, alterations in glutamate and GABA markers and signalling, and synaptic structural changes observed through MRI scans. On the other hand, MBP is associated with monoamine dysregulation and synaptic gain, evidenced by a heightened response to monoaminergic drugs, signs of autonomic dysregulation, and grey matter hypertrophy [3].
The specific characteristics of the stressor and individual predisposition contribute to the pattern of biological injury, determining whether MBP or ABP predominates and influences related behavioural responses. For instance, certain stressors, such as single prolonged stress, tend to mimic key symptoms of PTSD more closely. Additionally, different brain regions may exhibit distinct responses to specific stressors. For example, brief uncontrollable stress was found to induce synaptic loss in the infralimbic (medial prefrontal cortex involved in fear extinction) but not the prelimbic area (dorsal prefrontal cortex involved in fear acquisition) [3].
Importantly, the "vicious cycle" of CSP suggests that initial MBP-related behavioural disturbances further intensify the magnitude of stress, potentially leading to ABP and synaptic loss over time [3]. Therefore, the discrepancies observed in amygdala findings in PTSD (i.e., both hypertrophy and hypotrophy) may reflect the time course of the disorder, with chronic and severe PTSD being associated with more prominent ABP and synaptic loss [3].
Randox Clinical Studies Group, in association with the University of Ulster, Queens University, Belfast and the Medical University of South Carolina, has conducted research into PTSD, related sleep disturbances and potential biomarkers relating to the disorder, which may aid in risk stratification and diagnosis.
This review [26], published in Nature and Science of Sleep, identified 16 previously published investigations. This review highlighted that baroreceptor sensitivity was significantly lower in PTSD patients and showed deteriorations in the condition with declining sleep quality and amelioration of symptoms associated with improved sleep quality. This suggests sympathetic dominance in PTSD can be reduced by improving the sleep quality of the patient.
Data showed that increased ACTH levels observed in PTSD patients had a negative effect on their sleep quality, an association not made in healthy controls. Specifically, these changes in ACTH levels were found to be linked to a higher frequency of nocturnal awakenings in individuals with PTSD. Moreover, ACTH alterations were negatively associated with restorative slow-wave sleep and showed a negative correlation with delta-power sleep in PTSD patients.
This investigation showed that improvements in sleep quality resulted in changes in glucocorticoid regulatory mRNA expression, decreased pro-inflammatory cytokines and an increase in inflammatory regulatory mRNA, suggesting that disturbed sleep is a major cause of systemic inflammation in PTSD patients and further links the disorder with HPA axis dysregulation. Nevertheless, it remains unclear whether these sleep-related effects are a consequence of mechanisms aimed at protecting against excessive exposure to glucocorticoids or if the diminished sleep quality is a result of an improper activation of this mechanism. This improper activation may predispose individuals to the development of PTSD due to an attenuated glucocorticoid response to stress.
This review recommends that future treatment strategies should focus on improving sleep quality to help reduce the negative effects of PTSD, which should lead to a subsequent decrease in mortality rates related to PTSD, as well as conditions such as CVD, diabetes, and other metabolic disorders.
The objectives of this study [27] were to detect variances in the expression of several biomarkers that can be measured in peripheral blood samples between control subjects and individuals with PTSD. The study aimed to determine whether a single biomarker or a combination of biomarkers, when used in combination with clinical observations, could be utilised to assist in the identification of patients who are at risk of developing PTSD.
The authors identified 5 biomarkers, from a total of 37 measured, which were significantly altered in PTSD patients when compared with non-exposed controls, namely, HDL cholesterol, LDL cholesterol, epidermal growth factor (EGF), tissue plasminogen activator (tPA) and interleukin-8 (IL-8). However, subjects had not been asked to fast before their sample was drawn and therefore, HDL and LDL cholesterol were not included in subsequent analysis. The specificity and sensitivity data for the biomarkers EGF, IL-8 and tPA and the combination of the three are shown in Figure 4.
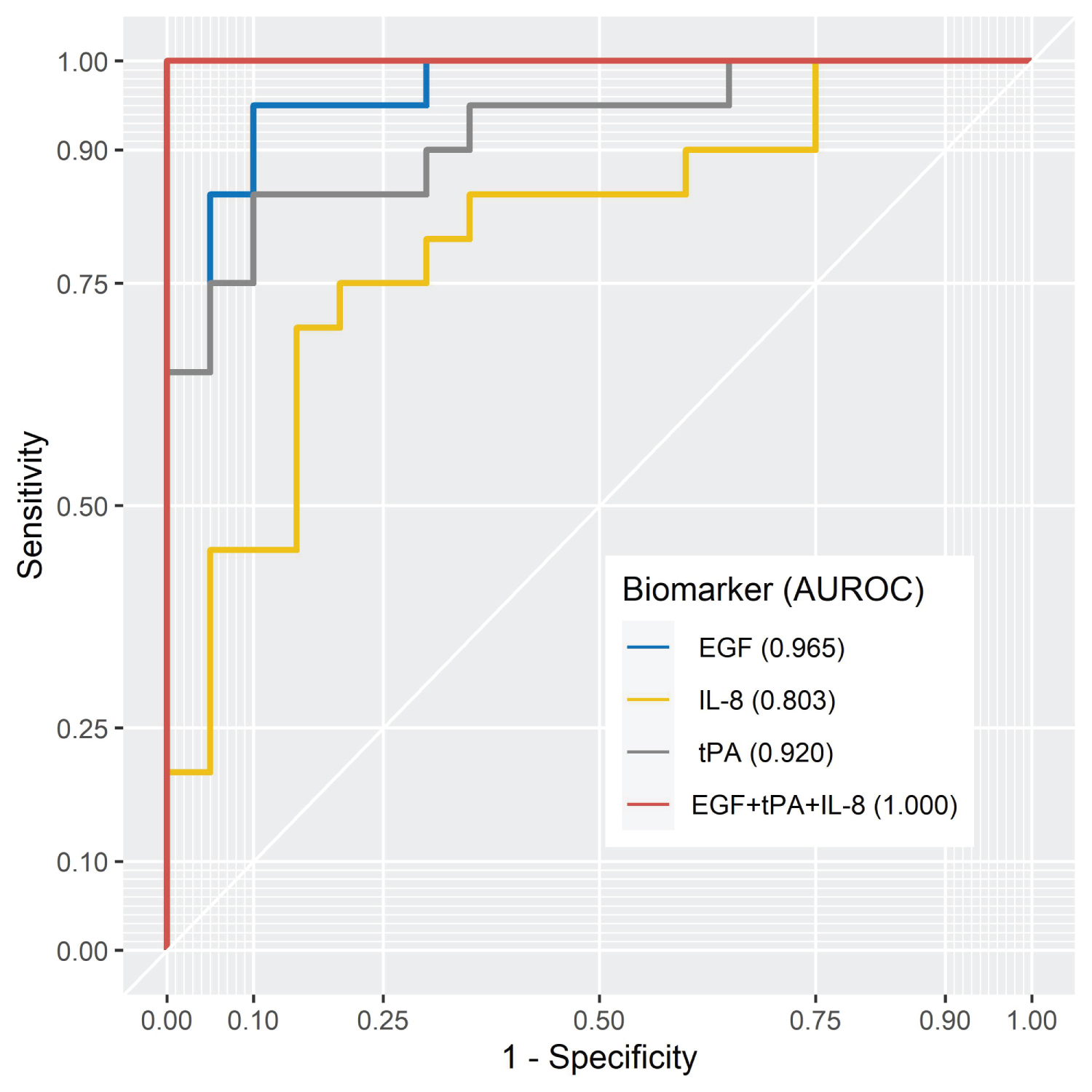 Figure 4: The AUROC curves for individual significant biomarkers and in combination [27].
View Figure 4
Figure 4: The AUROC curves for individual significant biomarkers and in combination [27].
View Figure 4
The data reported in this study show significantly lower levels of EGF and significantly elevated levels of tPA and IL-8 in PTSD patients when compared with non-exposed controls. The authors show that using EGF, IL-8 and tPA biomarkers in combination successfully differentiated control subjects from PTSD patients in all cases. In addition to the biomarker analysis, this study reported significant increases in diastolic blood pressure, increased incidence of substance abuse and increased comorbidity in PTSD patients compared to healthy controls. These data indicate a state of hyperarousal, which over sustained periods, is likely to result in exhaustion in the patient.
The biomarkers identified in this study may also provide a therapeutic strategy in which the alterations in the concentrations of the biomarkers may be reversed, improving the quality of sleep attained by PTSD patients. Measurement of EGF, IL-8 and tPA biomarkers offers a potential risk stratification approach to identify those most at risk of developing PTSD and other forms of mental illness.
The aim of the study [28] was to identify any differences in response to checklist questions provided by men and women. Clinicians conducted the interviews in a cohort recruited from California, USA and with controls matched for age, gender, and ethnicity.
The results highlighted a notable disparity, indicating that women had a threefold higher likelihood of experiencing sexual violence. However, the authors provide a caveat, noting that male participants exhibited hesitancy in disclosing or discussing numerous aspects pertaining to sexual violence or childhood sexual abuse that they might have encountered. Other than this disparity, no significant differences in the answers provided to the questionnaire were revealed based on gender. While the cohort in this study was small (N = 39) these data reiterate previous findings in the literature [29].
The objective of this pilot study [30] was to examine sociodemographic factors and comorbidities reported by individuals with PTSD, including sleep disturbances and chronic pain, and identify specific characteristics and behaviours that could be targeted for therapeutic interventions. The study sought to provide insights into potential avenues for effective, novel therapeutic approaches.
The results of this investigation showed significant differences in diastolic blood pressure between controls and PTSD patients. Common comorbidities encountered included arthritis, asthma, diabetes, and regular migraines, all of which were significantly more common among PTSD patients than in control subjects. In addition, common psychological comorbidities such as anxiety, ADD, bipolar disorder, depression, panic disorder and suicidal ideation were significantly more common in PTSD patients than in the control group.
The authors confirm their previous findings that sleep disturbances were more common in PTSD patients than in control subjects, 85% and 23% respectively. The data showed that 75% of PTSD patients in the study suffered chronic pain, most commonly in the lower back, neck and knee, a significant increase when compared to control subjects. However, no significant difference in the severity of pain was identified.
PTSD is a complex psychiatric condition with arcane pathological mechanisms. The symptoms of PTSD vary but commonly include intrusive memories, avoidance, negative mood, hyperarousal, and dissociative symptoms. The neurobiology of PTSD involves several interconnected brain regions, including the amygdala, hippocampus, prefrontal cortex, and neurotransmitter systems such as norepinephrine and GABA. Genetic and epigenetic factors also contribute to PTSD risk, with variations in genes related to stress response and neurotransmitter function influencing susceptibility. Epigenetic changes, such as DNA methylation, have been observed in PTSD patients and their descendants.
Several models have been proposed to explain the pathophysiology of PTSD, including the Synaptic Model of Chronic Stress Pathology CSP, the Network-based Model and the Dual Pathology Model. These models emphasize synaptic dysconnectivity, neuroinflammation, and alterations in neurotransmitter systems as key factors contributing to the development and maintenance of PTSD.
While the current diagnosis of PTSD relies on clinical assessment, there is a need for more objective methods of risk stratification and diagnosis. Biomarker research has identified specific biological biomarkers associated with PTSD risk, with the potential to improve early detection and personalized treatment approaches.
In conclusion, understanding the neurobiological mechanisms underlying PTSD is crucial for developing effective treatments and interventions. Biomarker analysis holds promise for enhancing the identification of individuals at risk and providing targeted interventions, ultimately improving the lives of those affected by this debilitating disorder. Further research is needed to validate these biomarkers and translate their use into clinical practice.
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Jason Armstrong, Joanne Watt, Jason Armstrong and Mark Ruddock are employees of Randox Laboratories Ltd but hold no shares in the Company. Peter Fitzgerald is the Managing Director of Randox Laboratories Ltd.