Primary or secondary hemophagocytic lymphohistiocytosis (HLH) is characterized by immune activation and life-threatening cytopenias. Causative relationship between a number of pathogens, autoimmune diseases and even hematologic malignancies with secondary HLH (sHLH) have been reported; however, correlation with myelodysplastic syndromes (MDS) is exceedingly unusual. HLH in COVID-19 positive patients has been described. Nevertheless, patients who have recovered after COVID-19 infection and test negative for the virus by RT-PCR may develop sHLH due to immune system dysregulation. We present here a rare case of sHLH manifestation after COVID-19 infection in a patient with a history of MDS. The recently revised H-score contributed to diagnosis.
COVID-19, Secondary hemophagocytic lymphohistiocytosis, Myelodysplastic syndrome, Sars-Cov-2, Bone marrow, Granuloma
Hemophagocytic lymphohistiocytosis (HLH) is a rare disorder induced by uncontrolled immune activation with a high mortality rate, that can affect all age groups [1-4]. According to the etiology, it is classified in two groups, namely primary or secondary. It remains an under-recognized entity. In such cases early diagnosis is critical [5]. The diagnosis of the syndrome is based on a series of clinical signs in combination with laboratory findings. The application of the recently revised diagnostic H-score helps to identify patients with HLH [6].
Many factors have been implicated in the development of the syndrome, including infections by fungi, protozoa, bacteria and viruses, autoimmune diseases and hematological or non-hematological malignancies [5]. At the same time, reports of myelodysplastic syndrome as a causal factor are rare [7].
Sars-Cov-2 is among the viruses that have been implicated as a causative agent of sHLH. However, there is a paucity of reported cases of sHLH in patients who have recovered from COVID-19 infection [8,9]. Here we report a case of a patient with a history of MDS, who had recovered from COVID-19 infection and developed sHLH.
A 71-years-old female patient with a history of MDS was transferred to the Hematology Department after 3 months of hospitalization in a COVID-19 Unit due to Sars-Cov-2 infection.
The diagnosis of MDS was made in June 2019 during a work-up for anemia (Hb:10g/dl) incidentally found on a routine blood test. Bone marrow aspiration and biopsy were significant for single lineage dysplasia of the erythroid series; cytogenetics disclosed trisomy 8. The patient was initially treated with recombinant human erythropoietin without response and soon she became transfusion dependent. Since March 2020, she received intermediate doses of corticosteroids and achieved transfusion independency sustained for over a year. In June 2021, an attempt of corticosteroid tapering led to transient worsening of anemia and need for transfusion. Anemia responded to increase of corticosteroid dose, and the patient remained transfusion independent until the emergence of Sars-Cov-2 infection.
At the time of her transfer to our Department, she had fully recovered from Sars Cov-2 infection, but was experiencing severe myopathy attributed to prolonged steroid treatment. On clinical examination, she had no organomegaly or other clinical findings, except of signs of iatrogenic Cushing’s syndrome. Complete blood counts showed mild anemia and thrombocytopenia: Hb: 11.2 g/dl, WBC: 5.600/mm3, PLT: 50.000/mm3.
Of note, the patient had been treated with high doses of corticosteroids during the previous two months. Soon after our attempt of corticosteroid tapering, a febrile episode (temperature over 40℃) occurred with a subsequent decrease in hemoglobin levels (Hb: 7.2 g/dl) accompanied by neutropenia (WBC: 1610/mm3, absolute neutrophil counts: 1400/mm3) and worsening of thrombocytopenia (PLT: 31.000/mm3). The patient received broad-spectrum antibiotics. Blood and urine cultures did not isolate any pathogens and computerized tomography of the chest excluded pulmonary involvement. No other site of infection was identified. At the same time, laboratory tests revealed elevated levels of serum triglycerides (365mg/dl) along with impressively high ferritin levels (63.737 ng/ml), while fibrinogen was normal (2.18 g/L). RT-PCR testing for Sars-Cov-2 was repeatedly negative during her hospitalization. Serology tests for human immunodeficiency virus, influenza A/B, cytomegalovirus (CMV) and Epstein-barr virus (EBV) were negative, while quantitative molecular testing for CMV and EBV viremia were negative. Due to the above findings and the high index of suspicion for HLH, we subsequentlyperformed a bone marrow aspiration. Bone marrow smears revealed hemophagocytosis (Figure 1). Bone marrow karyotype was normal (46,XX[20]) [7]. Trephine biopsy showed signs of erythroid dysplasia along with the presence of granulomas. However, the pathologists could not clearly identify morphological findings indicative of hemophagocytosis.
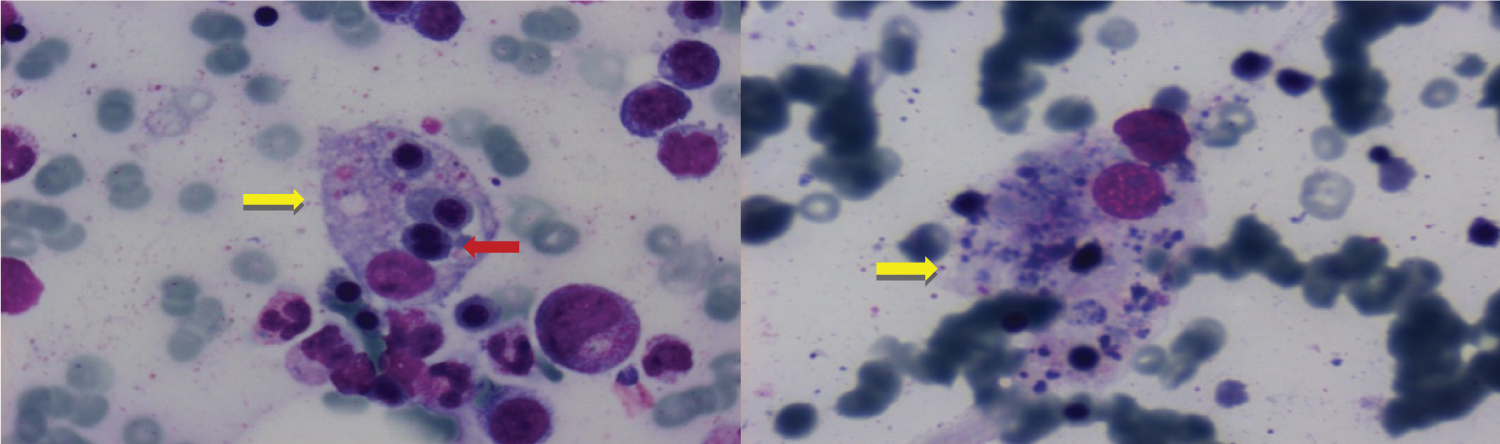 Figure 1: Bone marrow smears showing hemophagocytosis and signs of dysplasia of the erythroid lineage. Erythroid cells are phagocytized by histiocytic cells (yellow arrow). Nucleated red cell showing nuclear fragmentation (red arrow) with morphologic features consistent with dysplasia.
View Figure 1
Figure 1: Bone marrow smears showing hemophagocytosis and signs of dysplasia of the erythroid lineage. Erythroid cells are phagocytized by histiocytic cells (yellow arrow). Nucleated red cell showing nuclear fragmentation (red arrow) with morphologic features consistent with dysplasia.
View Figure 1
The revised H-score estimated for our case was 270 points; highlighting a probability of sHLH of > 99% (Table 1). The patient received treatment according to the HLH-94 protocol that includes etoposide. Response to treatment was notedwithin the following three weeks: patient’s ferritin levels remarkably dropped to 4.944 ng/ml, while at the same time she maintained stable levels of hemoglobin and platelet count. However, she developed neutropenia, that was attributed to etoposide toxicity. Unfortunately, the patient expired due to Acinetobacterbaumanii septicemia.
Table 1: Calculation of H-score-2014 in our case. View Table 1
The first case of HLH was reported in 1952 by Farquhar and Claireaux [10]. Its pathogenesis involves dysregulation of the immune system that leads to activation of cytotoxic T-lymphocytes and natural killer cells and increased production of cytokines resulting in activation of macrophages that is captured by the wide spectrum of hemophagocytic clinical and laboratory manifestations [11]. Αs an entity, it may affect patients of all age groups and a causal relationship between a number of factors and HLH has been reported, including viral infections (Herpes simplex virus (HSV), EBV, CMV, HIV, Influenza A and parvovirus B19), bacterial and fungal infection (Candida, Aspergillus), hematological malignancies and solid tumors, connective tissue disorders, immunodeficiency syndromes, and cellular therapies [12-14].
HLH is a rare and frequently unrecognized entity, while its diagnosis is challenging and often relies on clinical suspicion. Standardized diagnostic criteria are lacking, while hemophagocytosis as a morphologic finding on bone marrow smears can be observed in various conditions, such as malignancy, bone marrow aplasia post chemotherapy or even blood transfusion [8,15].
Patients with MDS rarely develop HLH. To our knowledge, our case is the fifth report of sHLH in patients with MDS. Recently, Sun et al described four cases of sHLH in patients with a history of MDS. In thiscase series, patients were older than 60 years with the exception of a pediatric one. Threeout of 4 (75%) harbored a karyotypic abnormality with trisomy 8 (+8) detected as a sole abnormality or within a complex karyotype, similarly to our case. One of them had refractory anemia with excess blasts and another one MDS with multi-lineage dysplasia. As for the remaining 2 cases, they were considered as intermediate and low risk MDS according to R-IPSS, with no further information given regarding WHO classification. Furthermore, in 75% of MDS-related sHLH published cases organomegaly was profound in clinical examination [7,16-18].
The H-Score takes into account clinical and laboratory findings with simplified numerical values, thus facilitating an early diagnosis of patients with Shlh [6,19,20]. For our case, the calculated H-score was 270 points, a result that predicted an over 99% probability of hemophagocytic syndrome. Thus, it became apparent to us, that sHLH was the most probable diagnosis in the context of a non-specific and complicated clinical scenario: our patient developed high fever and progressive cytopenias after being hospitalized for COVID-19 for 3 months, while being on steroids and multiple antibiotics for a protracted period of time. Of note, hemophagocytosis was morphologically evident on bone marrow smears, while it was inconspicuous on bone marrow sections. This observation highlights the importance of carefully inspecting smears, since single histiocytes phagocytosing cells may be easily missed.
sHLH has been associated with active Sars-Cov-2 infection, but reports in post-recovery patients with a negative molecular test are rare [8,13]. Overall, seven cases of post-COVID-19 HLH have been described in the literature. Median age was 40 years [range 2-69], with a male predominance (5 out of 7 cases). A serious underlying condition was reported in 2 out of 7, while in 4 cases past medical history was insignificant. Median time between the diagnosis of Sars-Cov-2 infection and HLH occurrence was 6 weeks [range 2-8]. Organomegaly was a prominent clinical finding, being reported in 57% of the patients. In all cases, H-score contributed to diagnosis. Five out of 7 patients were treated with corticosteroids while the remaining received corticosteroids in combination with etoposide [8,13,21-23]. The main characteristics, treatment and outcome of the aforementioned cases are presented in Supplementary Table 1. Although, no major differences in the clinical course between acute and post-COVID19 sHLH have been reported, treatment strategies seem to differ between these two groups. In acute infection, interleukin-1 receptor antagonists (anakinra) and interleukin-6 receptor antibodies (tocilizumab) have been used along with corticosteroids [24], while steroids with or without etoposide are more frequently applied in post-COVID19 sHLH.
Our case is the first post-COVID-19 HLH report in a patient with a clonal hematological malignancy. It is worth noting that the delay in the occurrence of the syndrome (12 weeks after the acute infection) can be possibly attributed to the high doses of corticosteroids that she had received in the COVID-19 Unit.
Immune-mediated macrophage activation is the hallmark of sHLH. Both Sars-Cov-2 infection [8,13,21-23,25] and MDS are associated with dysregulation of the immune system. In MDS, the association between cytotoxic T-lymphocytes and apoptosis has been implicated in the pathogenesis of refractory cytopenias. Moreover, autoimmune mechanisms are often associated with trisomy 8 MDS [26,27]. Further immune system dysfunction after Sars-Cov-2 infection may have contributed to the development of sHLH. However, we feel, that in our case, the recent Sars-Cov-2 infection, rather than MDS was the main trigger for HLH, since the patient’s underlying disease was well controlled with no signs of progression. The presence of granulomas in bone marrow sections further suggests a possible causative relationship between Sars-cov2 and sHLH, despite the fact that granulomas have not been reported as a histologic finding in trephine biopsies of patients affected by Sars-Cov-2 [28-31]. Granuloma formation involves the activation of macrophages by proinflammatory cytokines, such as TNF-α and interferon-γ due to a Th-1 response after several infectious and non-infectious agents. COVID-19 seems to produce similar inflammatory immune responses. Moreover the virus binds to the angiotensin-converting enzyme 2 (ACE-2) receptors and leads to activation and redistribution of immune cells [32-35]. There have been many references in the literature regarding the development of autoimmune diseases including granulomatous diseases as well as bone marrow involvement in patients following COVID-19 [36-42].
Sars-Cov-2 infection is a new disease entity. sHLH has been recognized as a severe complication during acute infection, but it should be kept in mind, that it may also arise in patients who have recovered from the infection, as part of the spectrum of late post-COVID-19 complications. Whether, this rare manifestation is more frequent in patients with underlying immune disturbances, needs to be further elucidated. The revised H-score is a valuable tool towards early diagnosis.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.