Lymphocytes are cells within the immune system that originate in the bone marrow (B lymphocytes) or thymus (T lymphocytes). It was thought that lymphocytes are not implied in the pathogenesis of vitiligo, but now we can confirm that T cell response rather than B cell humoral immune response is essential in the pathogenesis of vitiligo.
To further understand the role of lymphocytes in vitiligo.
Data were collected from the most recent and relevant manuscripts to clarify the role of lymphocytes in vitiligo with the most updated publications.
Melanocyte-specific, cytotoxic CD8+ T cells abundantly infiltrate active vitiligo lesions leading to the destruction of melanocytes. Specific cytotoxic T cells destroy targeted melanocytes by the release of Perforin, Granzyme, TNF-α, IFN-γ, and the Fas apoptotic proteins.
Several stimulatory and inhibitory molecules are available on lymphocytes and are in cross-talk with target cells including CTLA-4, TCR, PD-1, TIM-3, OX-40, CD40L, GITR and CD27. On the other hand, Treg are CD4+ CD25+ T cells that are controlling the autoimmunity and their function is abnormal in patients with vitiligo. Treg cells mediate the suppression of human melanocyte-specific CD8+ T cells through decreasing; proliferation, cytokine production, and T cell receptor affinity, and increasing susceptibility to apoptosis of the CD8+ T cells.
The immune destructive action of CD 8 lymphocytes is required to destroy melanocytes and induce vitiligo, hence immune-modulation is a cornerstone to halt disease extension.
Vitiligo, Lymphocytes, TReg, Immunity
TCR: T Cell Receptor; Th: T Helper Cell; APC: Antigen Presenting Cell; CTLA-4: Cytotoxic T- Lymphocyte Antigen 4; MHC: Major Histocompatibility Molecules; ICOS: Inducible T-Cell Costimulatory; PD-1: Programmed Death-1; PD1-L: Programmed Death-1 Ligand; GITR: Glucocorticoid-Induced TNF Receptor Family-Related Receptor; CTL: Cytotoxic T Cell; SV: Segmental Vitiligo; NSV: Non-Segmental Vitiligo; ROS: Reactive Oxygen Species; CCR7: C-C Chemokine Receptor Type 7; HLA: Human Leukocyte Antigen
The immune system possesses many types of cells including the lymphocytes of which two main sub-types are known to us, B lymphocytes and T lymphocytes. These lymphocytes originate in the bone marrow and then a group migrates to the thymus prenatally to mature. The cells rearrange their gene segments, eventually leading to the expression of a peculiar antigen-binding part, which is called the T-cell receptor (TCR). T cell receptor sequences are very diverse, with many possible sequence combinations. This antigen-binding component consists of two transmembrane molecules, the TCR-α and the TCR-β and are often the contact points with the antigenic peptides. This receptor allows lymphocytes to watch all cells in the body, ready to destroy cells suspected to pose harm. When matured lymphocytes leave the bone marrow and thymus, they are considered naive cells until exposed to their corresponding antigen [1-3].
The first exposure to self-antigens in the thymus results in the deletion of correlated lymphocytes to prevent the autoimmunity destructive process, while a second exposure to antigens for cells that were not deleted or escaped the thymic central deletion can change those naive cells into active cells [1,3,4].
The B cells are responsible mainly for the production of antibodies or induction of humoral immunity while T lymphocytes are responsible for cellular immunity and can be further classified into CD4+ and CD8+ cells. CD4+ cells are called T helper cells and can produce cytokines required for triggering the humoral and the cell-mediated immune responses. Different subsets are available including T helper cells 1, 2, and 17 (Th1, Th2, Th17), and TReg. On the other hand, CD8+ are called cytotoxic T cells and are responsible for direct cellular destruction in the adaptive immunity pathway (Figure 1) [1,5,6].
 Figure 1: Lymphocytes include T lymphocytes which develop in the thymus and B lymphocytes which develop in the bone marrow. T cells include CD4 helper cells and CD8 cytotoxic cells. Helper cells include Th1, Th2, Th17, and TReg cell sub-types.
View Figure 1
Figure 1: Lymphocytes include T lymphocytes which develop in the thymus and B lymphocytes which develop in the bone marrow. T cells include CD4 helper cells and CD8 cytotoxic cells. Helper cells include Th1, Th2, Th17, and TReg cell sub-types.
View Figure 1
Essential elements of the cellular immune response are the activation, clonal expansion, and differentiation of T cells. Activation of T Lymphocytes is divided into two phases; the first phase activates naive T cells and differentiates them into functional effector cells while in the second phase, these effector cells recognize antigen on specific target cells, which subsequently destroy the targeted cells. Antigens should be displayed on antigen-presenting cells (APCs) that are scanned by T-cells, and some of them will have TCRs that trigger activation of the T-cell and this stimulates the cell to divide producing daughter cells with similar TCR. This proliferation leads to it having thousands and even millions of those cells with the ability to function accordingly [1,2].
T cell activation requires at least two signals: i) TCR signal ii) Costimulatory signal (Figure 2). CD28 is the main co-stimulatory receptor and has two ligands, B7.1 (CD80) and B7.2 (CD86) which are expressed on APCs. The CD28 signals are essential for T cell activation, proliferation, and survival after T cell interaction with APCs. They control T cell survival by enhancing BCLXL expression, which prevents T cell death by apoptotic signals such as Fas activation or IL-2 inhibition. Activation of T cells in the absence of CD28 results in an anergic state [7,8].
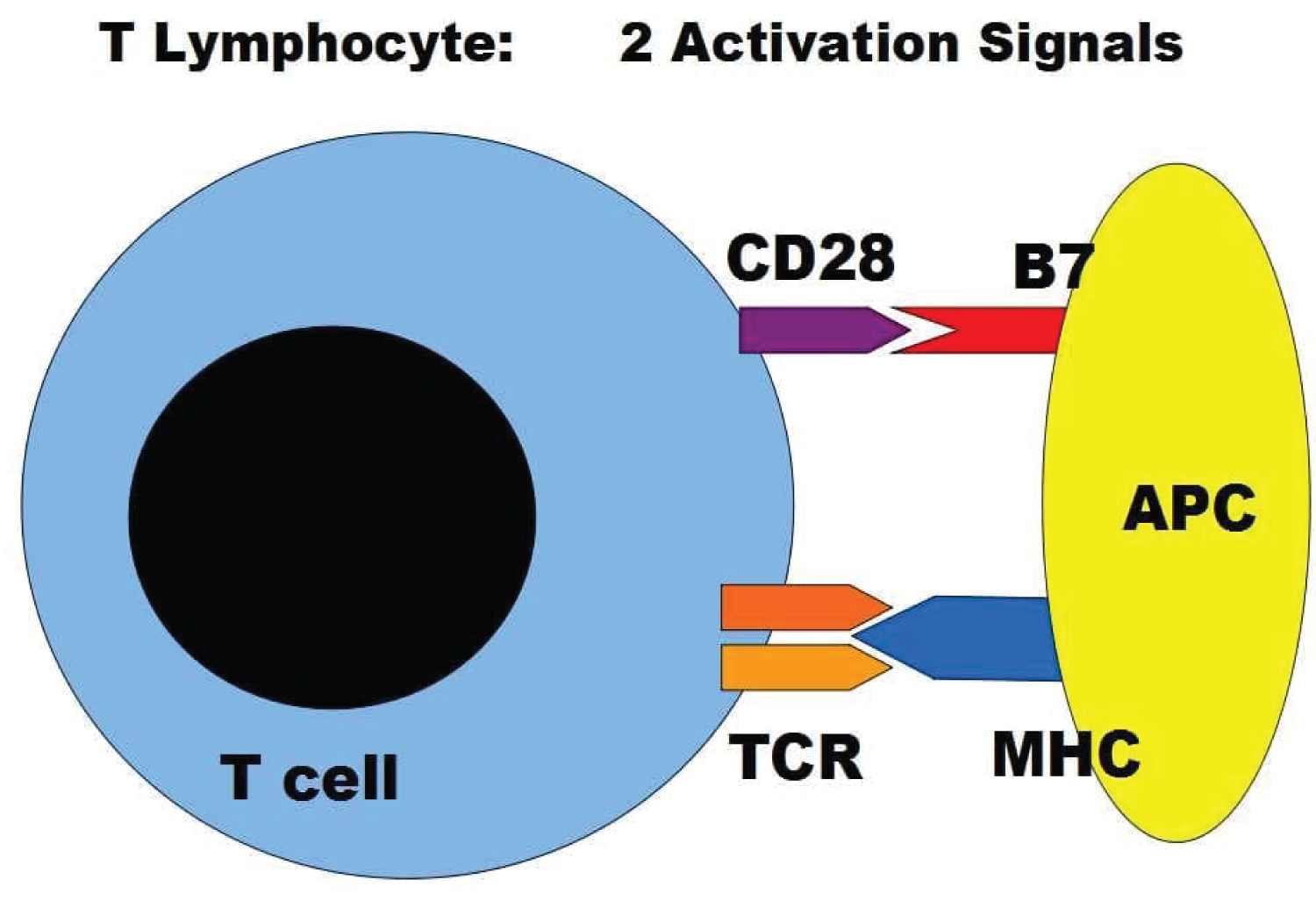 Figure 2: T lymphocytes need to be activated while encountering its specific antigen presented by antigen-presenting cell APC. Two signals are needed for this process of activation requiring the CD28 and TCR molecules on lymphocytes and the B7 and MHC molecules on the APC.
View Figure 2
Figure 2: T lymphocytes need to be activated while encountering its specific antigen presented by antigen-presenting cell APC. Two signals are needed for this process of activation requiring the CD28 and TCR molecules on lymphocytes and the B7 and MHC molecules on the APC.
View Figure 2
Science found several molecules on lymphocytes and APC/target cells in a complex cross-talk during antigen presentation and activation process including Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4), TCR, Major histocompatibility molecules (MHC), Inducible T-cell costimulator(ICOS), Programmed death-1 (PD-1), Programmed death-1 ligand (PD1-L), TIM-3, Galectin-9, OX-40, CD40, CD40L, glucocorticoid-induced TNF receptor family-related receptor (GITR), GITR-L, CD27 and others (Figure 3) [9].
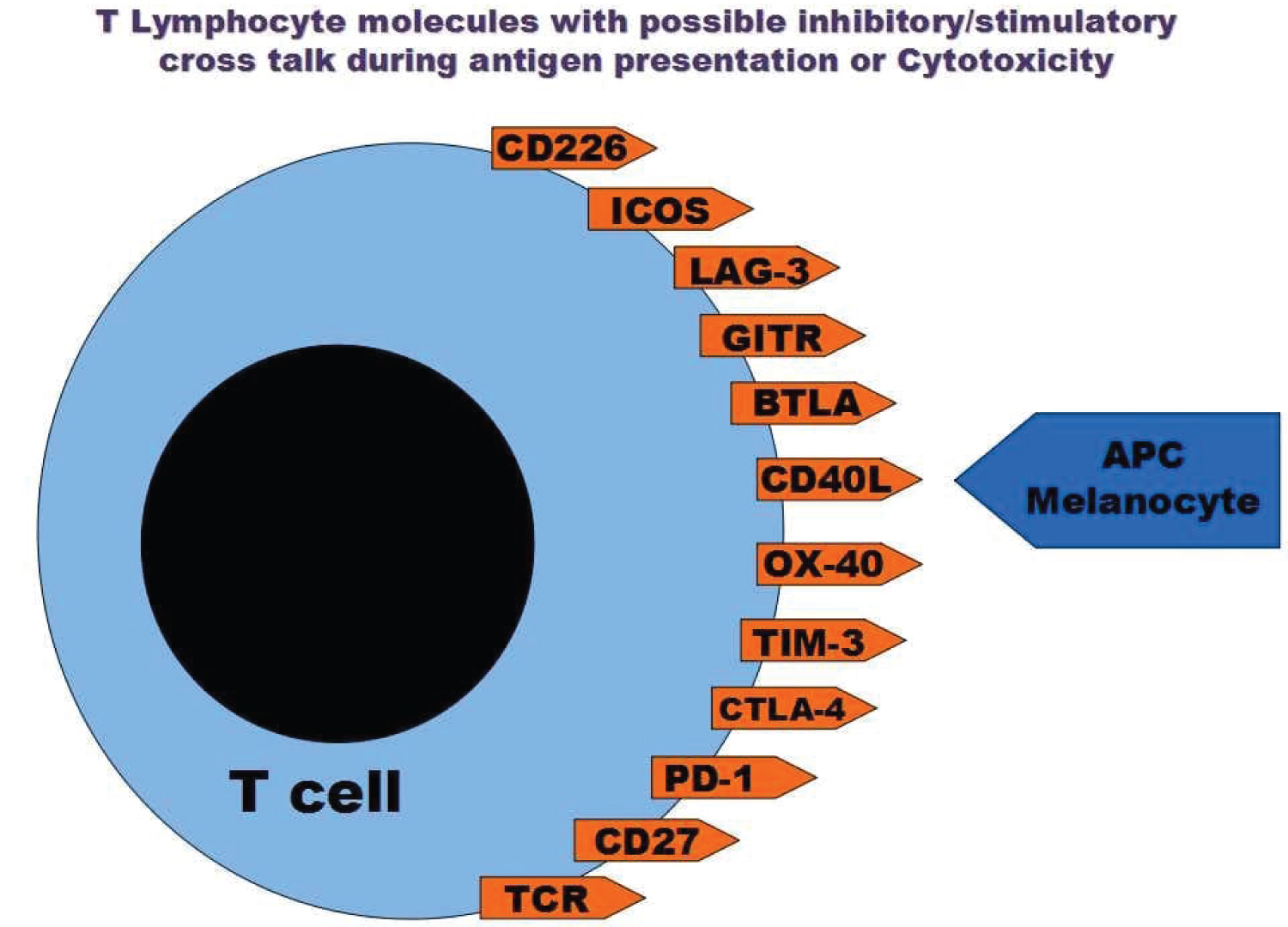 Figure 3: Several molecules were discovered on the lymphocytes that are in cross-talk with other cellular ligands to induce or inhibit the immune reaction.
View Figure 3
Figure 3: Several molecules were discovered on the lymphocytes that are in cross-talk with other cellular ligands to induce or inhibit the immune reaction.
View Figure 3
CTLA-4, a member of the immunoglobulin superfamily, was the first described inhibitory molecule and has a similar structure to CD28. Both molecules can bind to CD80 and CD86 on antigen-presenting cells. It was found that the lack of CTLA-4 can result in severe lymphoproliferative changes and fatality in rodents [10], as CTLA-4 can inhibit CD28 dependent T cell activation, cell cycle progression, and IL-2 production of T cells. It is clear that CTLA-4 inhibition is more pronounced after initiation of T cell activation and anti-CTLA-4 antibody activity is superior with continuous CD28 signaling, showing that upregulation of CTLA-4 is CD28 dependent [11,12].
CTLA-4 is a competitive antagonist of CD28 with a significantly higher binding affinity for the B7 molecules than that of CD28. Inhibitory effects of CTLA-4 were demonstrated in CD28 deficient mice showing that CTLA-4 can function also independently from CD28 [9].
So the existence of anergic clones of T cells is possibly due to failure or lacking costimulatory molecule B7 on APCs and the CTLA-4 inhibition of APC activation process [9,13].
After the successful presentation of the specific antigen epitope by APC, the activated T cells proliferate and differentiate further, and search for their targets with corresponding antigenic code to attack. The cytotoxic destructive process is carried out through:
• Secretion of cytokines, mainly TNF-α and IFN-γ.
• Production and release of cytotoxic granules, perforin, and granzymes. These are released only in the direction of the target cell, aligned along the immune synapse, to avoid unneeded damage to other healthy surrounding cells. CD8+ T cells can release their granules, kill a targeted cell, then move to another one and kill again acting as a serial killer.
• Express FasL on the cell surface, which binds to its receptor, Fas, on the surface of the target cell. These interactive molecules result in the activation of the caspase cascade, which subsequently leads to the apoptosis of the targeted cell. CD8+ T cells can express both molecules, Fas/FasL, and interaction of them can result in CD8+ T cells kill each other, a fratricide, to eliminate immune effector cells during the regression phase at the end of an immune reaction [6,14].
Other factors that can prevent the effector function of activated cytotoxic T cells (CTL) include; PD-1 inhibition of T cell destruction, TReg control of proliferation and activation of cytotoxic clones, Fas/FasL induction of apoptosis of CTL, lymphocytic and vascular endothelial cells, and other inhibitory elements [15,16].
A literature review of manuscripts, books, and peer-reviewed articles covering the topics of vitiligo and current understanding of lymphocyte function was performed by first author. Results from such studies were compiled and qualitatively appraised to create a coherent narrative of the current understanding of the role of lymphocytes in the pathology of vitiligo. This narrative was then edited by the authors to create a palatable discussion of the key points to serve as a learning resource for future researchers and fellow Immunologists and Dermatologists.
In a very early study in 1987 eminent authors described that lymphocytes are not incriminated in the pathogenesis of vitiligo. They studied lymphocyte subpopulations in patients with vitiligo and in controls with no significant differences in the results, of the then-available, immunological examinations, and according to their results, authors suggested that the lymphocytes are probably not implied in the pathogenesis of vitiligo [17].
Now it is agreed that Melanocyte-specific, cytotoxic CD8+ T cells are strongly implicated in the destruction of melanocytes. Patients with vitiligo have increasing numbers of cytotoxic CD8+ T cells in the blood compared with healthy individuals; these numbers correlate with disease activity. These CD8+ T cells when isolated from vitiligo patients can identify and kill normal human melanocytes in vitro. Furthermore, high numbers of CD8+ T cells were found in active lesions and proved necessary to kill the melanocytes. Infiltrating T cells isolated from vitiligo lesions show their ability to recognize melanocyte antigens, and when these cells were introduced in normally pigmented skin from the same patient, they migrated to the melanocytes and induced their apoptosis. CD8-depleted T cells could not kill melanocytes, whereas CD8-purified populations were more potent [6,18-21].
In the blood, it was found that the absolute and relative counts of lymphocyte subtypes are normal, yet the ratio of CD4+/CD8+ T-cells had an elevated median value in vitiligo cases. Probably due to domination of T-cell subtypes in the cutaneous site of autoimmune melanocyte loss [22].
Prenatally, thymic negative selection renders the developing T-cell repertoire tolerant to self-MHC/peptide ligands. The unusual large repertoire of CD8+ T cells specific for the Melan-A/MART-1 antigen has been confirmed and the reasons for its generation have remained mysterious [9,23].
It seems that first antigenic exposure was not properly performed as medullary thymic epithelial cells expressed a truncated Melan-A transcript, the product of misinitiation of transcription. Consequently, the protein product lacked the immunodominant epitope spanning residues [23,24], thus avoiding central tolerance to this antigen with subsequent production or escaping of this self CTLs.
Nevertheless, a significant proportion of the normal population showed detectable Melan-A tetramer+ CD8+ T cells in cord blood lymphocytes [25]. These cells were found in CD8+ thymocytes in thymuses from children. So, it appears that a high thymic output is responsible for the high frequency of these detected cells and is helped by a slow turnover of these cells during adult life [24].
In vitiligo, T cells were found to be expressing high levels of the skin-homing receptor cutaneous lymphocyte-associated antigen and their frequency is correlated with the extent of the depigmentation [18] and disease activity [19]. An affinity maturation develops later which facilitates the effective and specific anti-melanocytic activity [20,26].
Further searching for lymphocytes with possible TCR related to other melanocyte antigenic components, it was found that TRP1-specific cells were centrally deleted. Although sometimes TRP1-specific T cells may escape thymic deletion in certain conditions yet they are tolerant to TRP1 and do not induce vitiligo [27].
Another report showed the absence of tyrosinase-related protein-2/dopachrome tautomerase transcripts in peripheral blood mononuclear cells PBMCs from vitiligo patients, although transcripts were found in healthy individuals [28].
It seems that priming of T cells is not needed as memory cells here have well-known longevity characters. Even before their actual activation, they have shown a remarkable stability [29]. These lymphocytes with the specific TCR are ready to step forward and lymph node priming action is not of relevance in vitiligo yet direct presentation by melanocytes acting possibly as APCs is followed by the cytotoxic destruction process for themselves [29].
Now the cytotoxic lymphocytes with specific affinity to melanocytes are activated, but where are they heading? these are possible scenarios:
• Melanocytes with normal antigenic components (Autoantigens) like Melan-A or gp100 as a result of escaping of thymic clones, but possible excessive exposure to the antigenic elements may be needed. This is leading to episodes of depigmentation whenever an imbalance of tolerance develops [6].
• Mutated melanocytes are possible targets in genetically determined mosaic patterns in segmental vitiligo (SV) or some cases of nonsegmental vitiligo (NSV). This is leading to one attack of permanent and localized depigmentation early in life [30].
• Melanocytes carrying viral antigenic epitopes after viral infections. This is leading to episodes of depigmentation at sites of possibly infected cells. Viral infection was documented in many vitiligo cases and the frequencies of Melan-A-specific CD8+T cells in healthy individuals were found to be comparable to those measured of T cells specific for some viral epitopes [31].
• Newly developed antigenic components developing in melanocytes after trauma, infections, oxidative stress, or other harming elements. This is leading to episodes of depigmentation at sites of trauma or so. The accumulation of reactive oxygen species (ROS) in melanocytes will lead to melanocyte damage and the production of autoantigens. Cellular immunity targeting autoantigens takes an essential part in the destruction of melanocytes, with the help of heat shock protein 70, which eventually results in depigmentation [32].
• Melanocytes harboring shared antigenic components with other organs in cases of systemic autoimmune diseases. This is leading to depigmentation accompanied by other autoimmune diseases [33].
• Melanocytes with oncogenic antigens developing especially in cases with nevi. This may lead only to localized depigmentation in the form of a halo around the nevi [6,34].
In many situations, intrinsic defects in the melanocytes initiate the innate immunity reaction which subsequently leads to the initiation of the adaptive immunity reaction with macrophages and NK cells in action early in the course of the disease [21,35,36].
The macrophage-related factors like CD27, CD25, and MIF were checked in vitiligo and their levels were observed to be higher than that of controls with a greater correlation found for CD27 with disease activity. They are related to the innate immunity track and may confirm the need for this arm of action to activate the anergic clones. There is evidence that these markers have a capacity to indicate the probability of future disease progression [9,37]. These factors may ensure lymphocyte survival, T-cell proliferation, and memory cell formation and their expression supported helper Th1 development. They stimulate the CD4 helper cells and possibly TReg inhibition arm. CD25 was also found to be related to Th17 pathway of activation [38]. The increased number of circulating Th17 cells correlates with the extent of the disease [21], although more recent reports did not support a direct pathogenic role of IL-17 or Th17 cells in vitiligo at all [39].
Further studies of the immunophenotype of circulatory T-helper cells in patients with non-segmental vitiligo showed a negative correlation between the percentage of peripheral CD4+ CD25+ T cells (TReg) in vitiligo patients and disease activity according to VIDA score. There is a reduced percentage of CD4+ CD25+ T cells in peripheral blood of progressive vitiligo patients compared to the patients with stable vitiligo, and functional analysis of peripheral TRegs in vitiligo patients showed a correlation of TRegs function with the disease activity. Again, the percentage of circulating TReg cells increased significantly after treatment-induced disease stabilization [40,41].
TReg cell-mediated suppression of human melanocyte-specific CD8+ T cells in vitro causes a decline in proliferation, cytokine production, and T cell receptor affinity, and increases the susceptibility to apoptosis, expression of cell surface markers CC-chemokine receptor 7 (CCR7), and CTLA4 of the CD8+ T cells. However, this phenotype is only induced in melanocyte-specific CD8+ T cells from healthy subjects, but not in those from vitiligo cases, which suggests that TReg cells are indeed important in controlling the development of autoimmunity and that their function might be abnormal in patients with vitiligo [42,43].
So, in vitiligo, TReg cells are decreased or functionally defective, and they show deficient skin homing and low expression of transforming growth factor-β and CC-chemokine ligand 21. Reduced peripheral TReg cell numbers have been reported mainly in early age-of-onset patients (1-20 years) compared to those with late-onset vitiligo. Despite all of these, total consensus on changes and the dysfunction of TReg cells in vitiligo has not yet been fully established [13,21,29,44,45].
It was found that CD4 T cell-dependent autoimmunity against a melanocyte neoantigen induces spontaneous vitiligo and depends upon Fas-Fas ligand interactions. The destruction of melanocytes is partially inhibited by blocking Fas-Fas ligand interactions. This signifies the importance of local control of autoimmunity, as vitiligo in most cases, remains patchy and never proceeds to confluence even when T cells are abundant [46].
Genetic variants in the FAS gene were recorded in other researches concluding that they potentially affect the risk of vitiligo development [47].
As previously mentioned several molecules on lymphocytes and APC/Melanocytes are in a complex cross-talk (Figure 3) [9], but are they abnormal in vitiligo?, some reports described the absence of defective costimulatory signal or CD 40 and their levels were comparable between vitiligo cases and control subjects, referring to the lack of importance of these elements for the induction of vitiligo possibly as no further priming is required during disease activity as described in many other reports. CD40 ND CD40L were not changed between stable vitiligo and active cases [28,38].
CD28, CTLA4, and ICOS were studied too and although they are important regulators of the immune system yet in vitiligo, no polymorphism or changes were recorded to conclude that the CD28, CTLA4, and ICOS genes may not be associated with NSV [30,48]. A similar experiment was done on melanoma cases with the same outcome [49].
On the other hand, the expression of inhibitory factors PD-1 and Tim-3 and its ligand galectin-9 was found to be significantly elevated in vitiligo blood and skin and was also correlated with disease activity. An expected positive role in disease activity was claimed by many authors [50], although many other manuscripts described these elements as inhibitory rather than stimulatory factors. This increased expression of such inhibitory factors can be explained as being part of the exhaustion of T cells after fulfilling its destructive process during vitiligo activity. Their presence is a part of the existence of T cells and cannot be related to the halting process of the disease [51].
Another report that studied GITR family-related receptor on CD8 T cells confirmed that this can induce protective and high-avidity T cell responses as GITR stimulation-induced very weak CD8 T cell responses to melanocyte differentiation antigens expressed by the tumor and did not induce autoimmune vitiligo [52]. Many of these molecules were described as inhibitory elements for T cells, and so many investigators used their blocking factors during the management of tumors like melanoma to release the cytotoxic lymphocytes against the targeted tumor cells [9,37,50].
Although many factors of suppression were not reported to differ between vitiligo and control cases, yet blocking their actions during treatment of melanoma or other tumors led to the induction of vitiligo while improving the tumor status. This can confirm their persistent role in maintaining the equilibrium and the tolerance towards the melanocytes [53,54].
B lymphocytes are responsible for the production of antibodies that are in duty for eliminating harmful elements from the body, what we call humoral immunity.
The production of antimelanocyte antibodies is possibly elicited when there is a genetic predisposition to autoimmunity or cross-reacting antigens, expressed on cells or by infecting microorganisms. Again, antimelanocyte antibodies might result from a humoral immune response following the damage of the pigment cell [18-21].
Autoantibodies were found in the blood of vitiligo patients but these antibodies did not show affinity against the melanocyte differentiation antigen Melan-A. In many previous studies, the sera of patients with vitiligo did not show autoantibodies to the melanocyte-specific protein Melan-A. It is possible that Melan-A only contains epitops specific to induce cellular rather than humoral autoreactivity, or that their epitopes are not sufficiently exposed to the immune recognition system and so fail to stimulate an auto-antibody response, at least in the setting of vitiligo [55].
The presence of antibodies against melanocytes did not correlate with the disease activity or other relevant parameters, therefore screening for these antibodies does not appear to be clinically relevant [56]. It is conflicting that other reports declared the other point of view stating that serum antibodies to melanocytes in patients with vitiligo are predictors of disease progression [57], but now we can conclude that T cell response rather than a humoral immune response is important in the pathogenesis of vitiligo.
Most of the lymphocyte studies are dealing with NSV and what about SV cases?. In fact, there is an overlapping autoimmune mechanism between SV and NSV. T-cell inflammatory responses are present also in the active margin of SV lesions, Lymphocytic infiltration can be seen in many cases. CD4+ T cells infiltrated the dermis, while CD8+ T cells were present in the epidermis or attached to the basal layer early in the evolving disease. The increase in the number of CD8+ T cells was significant suggesting that SV also has an autoimmune mechanism. T cells are also involved in early evolving segmental vitiligo [48].
Neither precursor frequencies of Melan-A-specific T lymphocytes nor their status of activation differs significantly between vitiligo and melanoma cases. But a higher affinity of vitiligo T cells exists and only vitiligo T cells are capable of efficient receptor down-regulation and IFN-γ production in response to Human Leukocyte Antigen (HLA)-matched melanoma cells, suggesting that this difference in receptor affinity is of great relevance. It was claimed that Melan-A-specific cells circulating in melanoma patients are either ignorant or anergic. Vitiligo-like depigmentation sometimes occurring in patients with malignant melanoma, either spontaneously or more often as a result of successful immunotherapy, showing such dualistic immunity. So, there is a qualitative difference between the cytotoxic T lymphocyte responses to melanocyte antigens in melanoma and vitiligo and the immune response to Melan-A in vitiligo may be analogous to that of melanoma, in that cellular reactivity is predominant and a humoral response absent [26,55].
For the induction of the vitiligo battle, an army is needed with loaded weapons and orders to attack the enemy. The army composes of clones of lymphocytes produced in the thymus with specific TCR related to melanocytes. At first, these clones mature in the thymus and are released as naive members, then the enemy exposes itself, showing its suspected malicious nature which allows the induction of orders to produce more trained army members in thousands or even millions supplied with weapons, loaded and ready to fire. The malicious nature of the enemy is exposed after oxidative stress, infections, trauma, or by mutations in mosaic patterns of vitiligo whether SV or NSV, or even by oncogenic changes. The army heads to the enemy territories guided by its radar or the special affinity characters. Perforin, Granzyme, TNF-α, IFN-γ, and the Fas pathway of apoptosis are the weapons that are well-aimed and fired at the enemy destroying melanocytes and inducing depigmentation.
In melanoma cases, a similar army of lymphocytes is produced but without affinity for the enemy and with opposing orders not to attack and without loaded weapons. These opposing orders handed to the army of lymphocytes include PD-1, CTLA-4, Fas/Fas-L, and other inhibitory signals and the TReg cells are the usual correspondence soldiers. So the army here becomes a neutral army. These opposing inhibitory orders are abundant not only in melanoma cases but also in normal individuals, neutralizing the destructive power and hampering a possible war against melanocytes even after exposing its antigenic nature following trauma or other previously mentioned triggering factors.
The authors have no conflicts of interest to declare.
No funding source was used for this project.
S.A. was responsible for the literature search and writing of this manuscript; E.T. was responsible for proofreading, editing, and formatting the current manuscript.