Current treatment therapeutic approach for tuberculosis is the administration of first line drugs in the form of tablets and capsules for 4-6 months however; this approach leads to sever adverse effect. Therefore, present study was designed to achieving local and sustained targeting of ant tuberculosis drugs in order to reduce dose n frequency. Nanoparticles (NPs) have been found to be potential targeted and controlled release drug delivery systems. Various drugs can be loaded in the NPs to achieve targeted delivery. Nanoparticles of antituberculosis drugs are generally prepared by using chitosan and PLGA polymer. Nanoparticles of Chitosan being biodegradable, biocompatible, less toxic and easy to prepare, are an effective and potential tool for drug delivery. The selection of a nanoencapsulation technique should consider drug property, nanoparticle quality, scale-up feasibility, manufacturing costs, personnel safety, environmental impact, waste disposal, and the like. In recent years, there have been a plethora of nanoengineering approaches for the development of poly (lactide-co-glycolide) (PLGA) nanoparticulate carrier systems. However, overlooking the multifaceted issues in the preparation and characterization of PLGA-based nanoparticles. Relevant information might be helpful to those who prepare and develop nanoparticles of ant tuberculosis drugs that meet their specific demands.
Biodegradable, Drug delivery, Nanoparticles, Nanoencapsulation
In spite of the absolute number of incident TB cases falling globally, tuberculosis (TB) continues to be the leading cause of mortality worldwide and has also been considered to be an occupational disease in the health care setup [1]. One of the major problems in the current treatment of tuberculosis is the noncompliance to prescribed regimens, primarily because treatment of TB involves continuous, frequent multiple drug dosing. Adherence to treatment and the outcome of therapy could be improved with the introduction of long-duration drug formulations releasing the antitubercular agents in a slow and sustained manner [2]. Worldwide, around 10 million people fall ill with tuberculosis (TB) each year. TB is one of the top 10 causes of death, and the leading cause from a single infectious agent (Mycobacterium tuberculosis), ranking above HIV/AIDS. The disease can affect anyone anywhere, but most people who develop TB (about 90%) are adults, the male:female ratio is 2:1, and case rates at national level vary from less than 50 to more than 5000 per 1 million population per year. Almost 90% of cases each year are in 30 high TB burden countries. Globally, an estimated 1.7 billion people are infected with M. tuberculosis and are thus at risk of developing the disease [3]. With a timely diagnosis and treatment Tuberculosis (TB) is an infectious disease caused by Mycobacterium tuberculosis, a microorganism that usually attacks not only lungs but also other parts of the body such as spine, kidney, and brain [4].
The current antibiotic treatment of active TB consists in the administration of the first-line drugs isoniazid (INH), rifampin (RIF), pyrazinamide (PZA), and ethambutol (EMB) for 2 months, followed by RIF and INH for 4 months. Poor adherence to long-term therapy increases the number of patients harboring multidrug-resistant (MDR) M. tuberculosis strains (i.e., resistant at least to INH and RIF) and extensively drug-resistant strains (i.e., MDR strains resistant to any fluoroquinolone (FQ) and to at least one injectable second-line drug, kanamycin, amikacin (AK), or capreomycin (CP) [5,6].
Besides active TB, an estimated 2 billion people in the world have latent TB infection (LTBI) i.e., they harbor M. tuberculosis in a non-replicating (NR) (dormant) stage in their tissues, with 10% of persons reactivating to active TB lifetime [4]. As similar to active TB, the treatment of decades that latent TB is also very long, including 9 months of INH or 3 months of RFP plus INH [7].
During the two span the discovery of streptomycin in 1943 and that of rifampicin in 1963, practically all the active drugs against M. tuberculosis were discovered, making it very easy to cure TB [8,9].
Furthermore, during those years, the fundamental basis of TB treatment was defined based on the analysis of multiple randomized clinical trials (RCTs), the fundaments of which are still entirely valid today [10]: 1) Combining different effective drugs to avoid the selection of resistant M. tuberculosis strains; and 2) Ensuring that treatment is long enough to sterilize the tissues infected with M. tuberculosis and, therefore, prevent relapse [11,12].
It is currently accepted that any anti-TB treatment should include at least four drugs that are likely to be effective; of which at least two are essential or core drugs and two are companion drugs. The core drugs are those with the capacity to kill M. tuberculosis in any of its metabolic phases. In contrast, the role of the companion drugs is to accompany the essential core ones, protecting their action and avoiding resistance selection. Among the core drugs, one should have good bactericidal activity and the other should present good sterilizing activity. These two drugs should ideally be maintained for the whole treatment duration [13]. The bactericidal drugs quickly reduce the bulk of the rapid multiplying bacilli, decreasing infectiousness and avoiding the disease's progression. The sterilizing drugs attack the dormant and semi-dormant bacillus populations, allowing cure while preventing relapse. The drugs with the best sterilizing activity are those that may reduce the duration of the treatment. The two companion drugs are no longer necessary after bacteriological conversion [14].
To build an appropriate anti-TB regimen TB, WHO recommends a stepwise process based on five groups for MDR/XDR-of anti-TB drugs. The stepwise process leading to the adequate design of a regimen suitable to cure MDR/XDR-TB patients is summarized in Table 1.
Table 1: Stepwise process based on five groups for MDR/XDR-of anti-TB drugs. View Table 1
The choice of the drugs is based on their efficacy and toxicity, where group 1 includes first-line drugs and group 2-5 include SLDs. Group 5 includes the drugs with potentially limited efficacy or limited clinical evidence.
Length of TB treatment:- Minimum length of treatment is 21 months, divided as follows: Intensive phase: 6 months and ≥ 4 months after culture conversion; longer if three effective drugs are not available during the continuation phase Continuation phase [15,16].
Group 1
All potentially effective group 1 drugs should be included in the regimen, considering that isoniazid, rifampicin and pyrazinamide are core drugs, and ethambutol is a companion drug. High-dose isoniazid should be added to an MDR/XDR-TB regimen when the katG mutation is not documented by the Genotype line probe assay (Quest Diagnostics, Madison, NJ, USA) but it should not be counted as one of the four active drugs. Pyrazinamide should always be used, although its drug susceptibility test is unreliable, but it also should not be considered as one of the four active drugs [8,17].
Group 2
According to the current WHO classification, group 2 includes the injectable SLDs and group 3 includes the fluoroquinolones. The fluoroquinolones (particularly the later-generation fluoroquinolones, like high-dose levofloxacin or moxifloxacin [7] are core drugs, with bactericidal and sterilizing activity, as well as a good safety profile, and their use predicts a favorable outcome in MDR-TB treatment. Conversely, the injectable SLDs have only bactericidal activity (not sterilizing) and their safety profile is clearly worse [9,10].
Group 3
If we include the fluoroquinolones in group 2, the question is what drugs should comprise group 3. Theoretically, they should be the injectable SLDs (the other currently used core drugs with bactericidal activity) [11,12]. However, considering future scenarios, to be confirmed by stronger evidence on efficacy and safety, group 3 should include three core oral drugs, linezolid, bedaquiline and delamanid, instead of the injectable. These three drugs might be able to change the bleak prognosis of MDR-TB patients with resistance to fluoroquinolones (some clinicians call these cases pre-XDR, using a non-approved definition [18,19].
Group 4
Following the proposed reclassification of groups 2 and 3, the injectable SLDs might have the characteristics of a future group 4 (Table 2). In fact, based on their bactericidal activity, they remain core SLDs but given their cumulative toxicity (more than 6-8 months of treatment increase the chance of deafness or kidney problems) and the need for parenteral administration, they would rank lower than the previously described compounds [20].
Table 2: List of advantages and drawbacks of chitosan nanoparticulate dosage form. View Table 2
Group 5
A future hypothetical group 5 should include the thionamides (ethionamide or prothionamide) that currently belong to group 4, of which they represent the best drugs: the only ones with some bactericidal activity [21]. Among the limitations of the thionamides are poor gastric tolerance, possible cross-resistance with isoniazid and lack of clinical trials analyzing their authentic role in the treatment of TB.
Over the past few years, the budding use of nanotechnology based therapy has been researched for replacing the administration of antibiotics or other drugs in the free form with an access using drugs that are encapsulated with nanoparticle [22].
Nanobead delivery has a remarkable feature of slow, sustained, and controlled release from a biodegradable particle. Different animal models have been tried out to develop an antibiotic therapy based on polymer technology against M. tuberculosis but unfortunately not a single model stands upto the expectation or imitates all the features of human TB [23].
The sizes of nanoparticles which are defined as submicron (< 1 um) colloidal particles are used as drug delivery vehicles. For therapeutic purposes, drugs can be covalently embedded to the particle surface or can be incorporated in the matrix of the particle. Nanoparticles comprise biocompatible and biodegradable materials such as polymers, which can be either natural (e.g., gelatin and albumin), synthetic (e.g., polylactides and polyalkylcyanoacrylates), or solid lipids (SLNR and NLCR) [24]. Nanoparticles are taken up more efficiently by cells than larger molecules which make them a promising transport and delivery system. These carriers are adapted to enable controlled, slow, and persistent drug release from the matrix [25]. Several methods have been discussed explicitly in several reviews for preparation and characterization of nanoparticles. Below are the advantages of nanoparticle-based drug delivery system for treatment of tuberculosis:
1) High constancy/longer time period,
2) High carrier ability; that is, multiple drugs can be encapsulated in the matrix,
3) Less side effects compared to conventional drugs,
4) Increased bioavailability (slow, sustained, and controlled drug release),
5) Viability of various routes of administration like oral delivery and inhalation,
6) Minimal side effects and improved compliance.
Polymeric nanoparticles have been synthesized by several methods depending on the requirements of their application and the physicochemical characteristics of the drug. The choice of the most suitable method plays a vital role in order to obtain PNPs with the desired properties for a particular application [26].
Water-in-Oil (w/o) emulsion phase separation: An aqueous drug solution is emulsified in an organic solution (e.g., methylene chloride) in which PLGA is dissolved. A phase inducer (e.g., silicone oil, vegetable oil, or mineral oil), being an anti-solvent for PLGA but a solvent for the organic solvent, is added to the w/o emulsion to induce coacervation. Formation of coacervates around the inner aqueous phase and their fusion generate embryonic PLGA particulates. To harden them, the suspension is further treated with an organic solvent (e.g., heptane), which is a non-solvent for PLGA but a solvent for the dispersed solvent and the phase inducer. EP 2131815 describes the principle of this process and its application in encapsulation of various water-soluble drugs into PLGA particles [27]. This method provides high encapsulation efficiencies toward hydrophilic drugs, due to their insolubility in organic solvents. However, controlling a coacervation step is quite difficult, and this method requires high costs of handling oils in large quantities [28].
Oil-in-Water (o/w) emulsion solvent evaporation/extraction: An o/w emulsion solvent evaporation method involves the following steps: (i) Emulsification of a polymeric dispersed phase in an emulsifier-containing aqueous phase; (ii) Solvent diffusion from emulsion droplets into the aqueous phase; (iii) Solvent removal by evaporation; and (iv) Solidification of emulsion droplets into PLGA nanoparticles. Solvent evaporation is usually carried out by continuous stirring at ambient pressure, but its rate can be accelerated under reduced pressure, vacuum, or elevated temperature [29]. Methylene chloride, which has a low boiling point of 39.8 ℃ and negligible water solubility (1.32 wt %), is the preferred solvent of choice. This technique is particularly useful for loading hydrophobic drugs into PLGA nanoparticles. Prior to emulsification, an emulsifier is added to a continuous phase and/or a PLGA organic solution. Common examples of surfactants and/or stabilizers are polyvinyl alcohol (PVA), didodecyldimethylammonium bromide, polyvinyl pyrrolidone, solutol, polysorbate, poloxamer, carbopol, polyethylene glycol (PEG), sodium dodecyl sulfate, proteins, carbohydrates, lecithin, and PEG-lipid (e.g., PEG-ceramide, d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS). It is important to notice that the type and concentration of surfactant affect the nanoparticle quality, drug encapsulation efficiency, drug release, pharmacokinetics, and cellular uptake/interaction. Therefore, a surfactant should be selected in consideration of the intended functionality and quality of PLGA nanoparticles. In the practice of solvent extraction, an excessive amount of a quench liquid (i.e., water) is added into the o/w emulsion, in order to facilitate the quenching of the dispersed organic solvent into the aqueous phase. The amount of water used for solvent extraction usually exceeds at least 10 times the theoretical amount required for saturating water with the organic solvent. Ethyl acetate with a significant water miscibility (∼8 wt %) is frequently used with this nanoencapsulation method. Solvent evaporation is often hybridized with solvent extraction to effectively remove organic solvents. Other treatments (e.g., dialysis, diafiltration/ultrafiltration, vacuum, and/or cosolvent treatment) are performed too. Table 1 summarizes experimental conditions used for nanoencapsulation of hydrophobic drugs and the major properties of PLGA nanoparticles. Important process parameters are PLGA, solvent, drug concentration, drug-polymer ratio, surfactant/emulsifier, mixing device, organic phase-continuous phase ratio, solvent removal (method and rate), and freeze-drying condition (method and lyoprotectant) [19,30-32].
Oil-in-Oil (o1/o2) emulsion solvent evaporation/extraction: A PLGA/drug/organic solvent dispersed phase (o1) is emulsified in a continuous oil phase (e.g., silicone oil, liquid paraffin, or vegetable oil; o2), and the process of solvent removal is carried out as mentioned earlier. Following this principle, Mahdavi, et al. emulsified a PLGA/acetonitrile dispersed phase (o1) in mineral oil (o2) [33]. After solvent removal, PLGA nanoparticles were recovered by centrifugation and washed by n-hexane to eliminate residual mineral oil. The mean diameters and production yield of their nanoparticles were 640∼890 nm and 38.5∼45.3%, respectively. Kashi, et al. dissolved PLGA and minocycline in acetonitrile and emulsified the dispersed phase in Span 80-containing mineral oil [34]. The resulting nano-emulsion was continuously stirred in 55 ℃ for 2 hr to remove acetonitrile by evaporation. Nanoparticles were collected by centrifugation and washed with n-hexane.
Oil-in-Water (o/w) salting-out technique: This technique involves the emulsification of a polymeric, drug containing organic solution into an electrolyte-saturated aqueous solution. A water-miscible organic solvent is used as a dispersed solvent for PLGA [35]. When a polymeric dispersed phase is mixed with an aqueous phase saturated with electrolytes, the salting-out effect prevents the mixing of the organic solvent with water, thereby producing a stable o/w emulsion. A reverse salting-out effect is induced by adding an excessive amount of water to this emulsion. This triggers the diffusion of the water-miscible solvent out of emulsion droplets into the aqueous phase, which leads to formation of nanoparticles. Examples of electrolytes used in the salting-out process are magnesium acetate, magnesium chloride, calcium chloride, and sodium chloride. Benefits of this nanoencapsulation method include the use of non-halogenated solvents to dissolve PLGA.
Water-in-Oil-in-Water (w1/o/w2) emulsion solvent evaporation/extraction: This method is used to encapsulate hydrophilic drugs into PLGA nanoparticles. A hydrophilic drug is dissolved in water (w1), while PLGA is dissolved in Moderate encapsulation efficiency of 51.2% was attained with this nanoencapsulation method [36,37].
When oil dissolved in a water-miscible organic solvent is mixed with a large amount of water, spontaneous emulsification takes place to make a kinetically stable nanoemulsion. This so-called Ouzo effect is a process by which hydrophobic solutes are dispersed in water as Nano sized droplets or particles [38]. When the Ouzo emulsification process is applied for preparing polymeric nanoparticles, the nanoencapsulation method is referred to as nanoprecipitation. In this practice, a hydrophobic polymer material is first dissolved in a water miscible solvent. This polymeric dispersed phase is mixed with an aqueous phase by use of a low-energy mixing device. The solution resulting from the mixing of the polymeric phase with anti-solvent (i.e., water) is a non-solvent for the polymer solutes. As a consequence, hydrophobic polymer solutes become aggregated to form Nano sized particles. When Fessi, et al. used this nano-precipitation technique to prepare PLGA nanoparticles, they named it as the solvent displacement method [39,40].
Strictly speaking, nanoprecipitation is a single-step formation of polymeric dispersion in water. In the field of the preparation of PLGA nanoparticles, this method has been slightly modified to include the steps of emulsion formation and nanoparticle hardening. Based on this strategy, Niwa, et al. proposed the so-called SESD technique [41,42]. Its key strategy was to use a binary solvent mixture consisting of water-immiscible and water-miscible solvents (e.g., methylene chloride-acetone). Ibrahim, et al. also used a methylene chloride-acetone binary system to prepare celecoxib-loaded. PLGA nanoparticles. Acetone, which is miscible with methylene chloride and water, helps decrease the interfacial tension between methylene chloride and water [43]. Additionally, the interfacial turbulence caused by the rapid diffusion of acetone from emulsion droplets to water contribute to the spontaneous breakups of emulsion droplets into Nano sized ones. Solvent evaporation follows to remove methylene chloride from the Nanoemulsion, so as to produce solid PLGA nanoparticles. These are the underlying principles of the SESD technique. On the basis of these considerations, SESD can be regarded as a hybrid of emulsion solvent evaporation/extraction and Nanoprecipitation. There are other reports that exploit the importance of a solvent system for manipulating the physicochemical attributes of resultant nanoparticles [44,45].
Nakashima, et al. proposed the shirasu porous glass (SPG) membrane-emulsification technique to prepare various kinds of particulate matters [46,47]. Since then, this technique has been widely explored for the preparation of liposomes, micro particles, and Nanoemulsions [48]. Briefly, this emulsification system forces a polymeric dispersed phase to permeate through a porous membrane into an aqueous continuous phase. The resultant emulsion is subject to the process of solvent removal, thereby generating nanoparticles. The membrane properties (pore size, spatial distribution, and tortuosity) play a decisive role on the size of the emulsion droplets and/or nanoparticles. This feature is distinctive from other emulsification systems using high-energy sonicators, high-shear rotor/stator homogenizers, or high-pressure micro fluidizers. While these conventional mechanical methods require high energy input (106-108 J⋅m-3), the membrane emulsification method uses relatively low energy input (104-106 J⋅m-3). For membrane emulsification, either a polymeric dispersed phase or a premix (i.e., a ready-made coarse emulsion) is passed through a membrane. For example, Wei, et al. emulsified a PLGA dispersed phase in an aqueous phase by using a low-speed rotor/stator homogenizer [49]. This coarse emulsion had large droplet sizes with a broad size distribution. By extruding the premix through a SPG membrane (1.2∼9 µm pore size) by use of nitrogen gas pressure, they were able to produce uniform PLGA nanoparticles. Yu, et al. also encapsulated docetaxel into PLGA-TPGS nanoparticles by combining preemulsification and SPG membrane emulsification [50].
A number of attempts have been made to increase the loading efficiency of hydrophilic drugs into PLGA nanoparticles. One relevant strategy is to enhance the lip solubility of water-soluble drugs by use of a hydrophobic ion pairing (HIP) technique [51]. Briefly, bis (2-hylhexyl) sulfosuccinate sodium salt (AOT) was dissolved in methylene chloride, which was then vigorously stirred in an aqueous gentamicin sulfate solution. This caused a complexation reaction between the anionic AOT and the cationic gentamicin. The hydrophobic ionic complex, after being recovered from the organic phase by evaporation, was used for an o/w emulsion Nanoencapsulation process [52].
In this method, an aqueous solution of chitosan is added to an organic solvent containing a surfactant. Agitation is done simultaneously. Water is added to maintain the mixture in an optically transparent microemulsion phase. The amount of water is increased to obtain NPs of larger size as shown in Figure 1. Chitosan NPs prepared by this method have been used for encapsulation of doxorubicindextran conjugate [37].
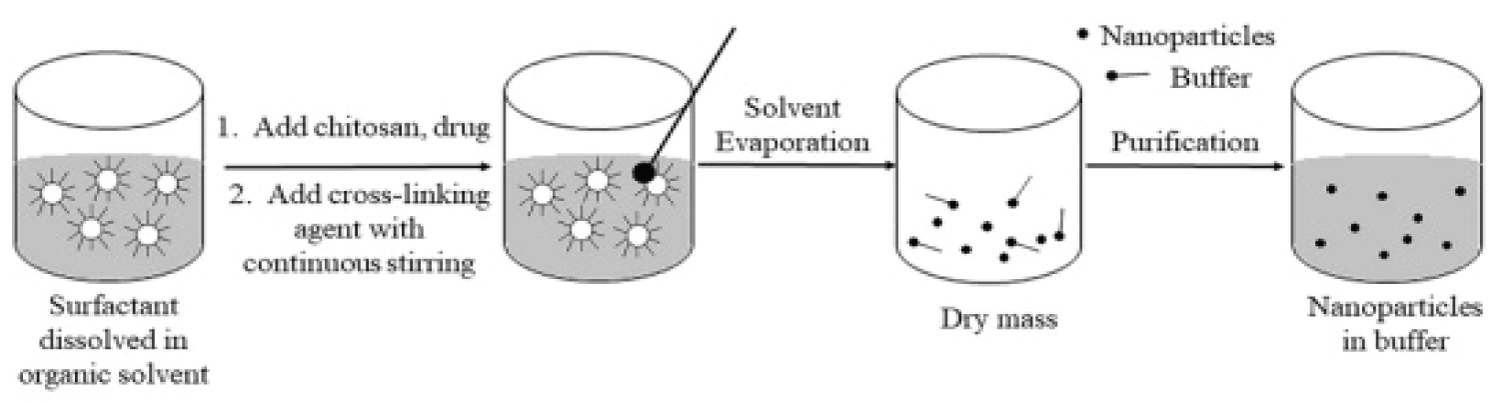 Figure 1: Diagrammatic representation of chitosan nanoparticles preparation by Reverse Micellar Method.
View Figure 1
Figure 1: Diagrammatic representation of chitosan nanoparticles preparation by Reverse Micellar Method.
View Figure 1
In this method ionic cross-linking is achieved by aggregation of chitosan or its derivatives with oppositely charged macromolecules or in the presence of ionic crosslinking agent. Tripolyphosphate is the most commonly used cross-linking agent. There is a formation of gels due to ionic linkage, therefore this method is also known as ionic-gelation method [53,54] (Figure 2).
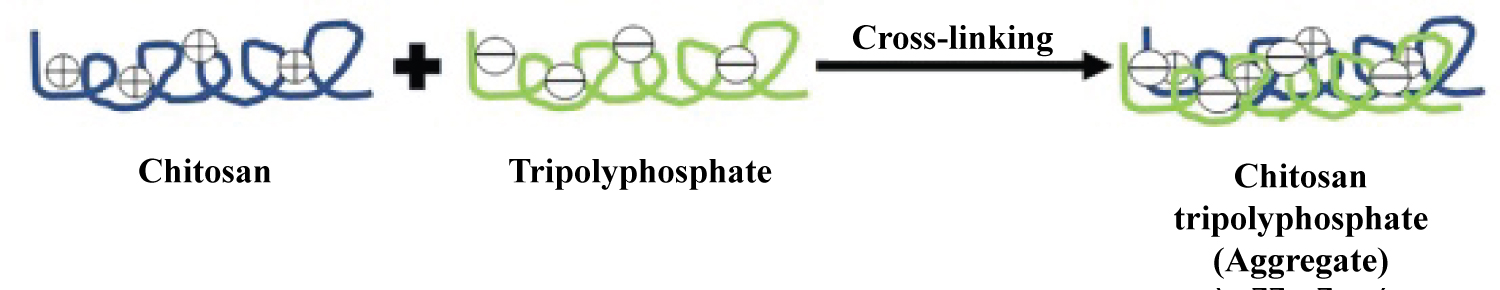 Figure 2: Diagrammatic representation of chitosan nanoparticles preparation by ionic cross-linking technique.
View Figure 2
Figure 2: Diagrammatic representation of chitosan nanoparticles preparation by ionic cross-linking technique.
View Figure 2
In this method there is formation of covalent bonds between chitosan or its derivatives and the functional cross-linking agent. Commonly used agents include polyethylene glycol, glutaraldehyde or monofunctional agents [55].
In this method, an aqueous solution of chitosan is added to an organic solvent containing a surfactant. Agitation is done simultaneously. Water is added to maintain the mixture in an optically transparent microemulsion phase. The amount of water is increased to obtain NPs of larger size [56,57].
This method was developed by Tokumitsu, et al. It is based on the principle of emulsion cross-linking and precipitation. In this method, two emulsions are prepared, one containing aqueous solution of chitosan and drug in liquid paraffin oil and the other containing aqueous chitosan solution of NaOH in liquid paraffin oil. Both the emulsions are mixed under high speed stirring and there will be formation of droplets which would collide and coalesce, thereby precipitating chitosan droplets to give small size particles. This method was used to prepare gadopentetic acid-loaded chitosan NPs [58].
Nanoparticles are generally characterized by their size, morphology and surface charge, using different techniques.
There are several tools for determining the size of nanoparticles as discussed below.
Dynamic light scattering: One technique that is widely used in size determination of PNPs in solution is dynamic light scattering (DLS), which is also known as photon correlation spectroscopy (PCS). In a DLS experiment, a colloidal suspension is illuminated by a monochromatic laser light that is scattered into a photon detector [59]. Due to the Brownian motion of the particles, the detected scattered light intensity fluctuates in time, and this is related to the particle size with the help of an autocorrelation function [60]. Based on the Stokes-Einstein equation the hydrodynamic diameter can be obtained. For spherical particles the relation is as follows:
where Dh is the hydrodynamic diameter, η is the relative viscosity of the solvent, kB is the Boltzmann's constant, T is the temperature and Dt is the translational diffusion coefficient (this is what is measured by DLS). One of the major advantages of DLS is that it provides information about the whole particulate population in short experiment duration. However, it has limited utility in determining the particle shape because DLS presumes all the particles being spherical in nature. Furthermore, as larger particles scatter more light than smaller ones, even small amounts of aggregates or dust particles could shift the particle size distribution to larger values. Therefore, one should be cautious when interpreting size information from DLS experiments. The nanoparticle size reported is usually the intensity-based diameter (Z-average) which is the value obtained directly from the correlation function. This can be converted by Mie theory to a volume or number distribution if there is no error in the intensity distribution. When comparing size results obtained by microscopy techniques, as these are number based metrologies, the corresponding size distribution for comparison is the number distribution. The DLS results should be supplemented with an imaging technique like AFM or SEM, which is helpful in resolving ambiguities associated with both techniques. For example, Sant and coworkers prepared PEG-g-PLA nanoparticles by the emulsification solvent evaporation method [61].
The size and morphology of nanocarriers exert a profound influence on the physical and chemical properties that determine their interaction with the environment and biological systems. There are certain techniques to analyze the morphology of nanoparticles. Microscopic techniques like SEM, TEM and AFM, along with particle size also determine other parameters like morphology or surface roughness of the nanoparticles [62].
Scanning electron microscope: For SEM characterization, nanoparticles dispersion should be first converted into a dry powder, which is then mounted on a sample holder followed by coating with a conductive metal, such as gold, using a sputter coater. The sample is then scanned with a focused fine beam of electrons. The surface characteristics of the sample are obtained from the secondary electrons emitted from the sample surface. The nanoparticles must be able to withstand vacuum, and the electron beam can damage the polymer [63].
Transmission electron microscope: TEM operates on different principles than SEM, yet it often brings the same type of data. The nanoparticles dispersion is deposited onto support grids or films. To make nanoparticles withstand the instrument vacuum and facilitate handling, they are fixed using either a negative staining material, such as phosphotungstic acid or derivatives, uranyl acetate, etc., or by plastic embedding. Alternative method is to expose the sample to liquid nitrogen temperatures after embedding in vitreous ice [64]. The surface characteristics of the sample are obtained when a beam of electrons is transmitted through an ultra thin sample, interacting with the sample as it passes through.
Atomic force microscopy: AFM is yet another tool used to characterize variety of surfaces, including nanoparticles, at the atomic level and it is one of the primary forms of scanning probe microscopes [65]. The prime advantage of AFM is its ability to image non-conducting samples without any specific treatment, thus allowing imaging of delicate biological and polymeric nano and microstructures. AFM requires minimal sample preparation and can be performed in ambient conditions [66]. Scanning with a sharp probe across its surface and then monitoring and compiling the tip-sample interactions provide the images of the sample surface.
The colloidal stability is analyzed through zeta potential of nanocarriers. This potential is an indirect measure of the charge. It corresponds to the potential difference between the outer Helmholtz plane and the surface of shear. Laser Doppler anemometry is the technique used to measure the zeta potential. It is based on the evaluation of the velocity of particles by the shift caused in the interference fringe, which is produced by the intersection of two laser beams. The electrophoresis mobility is then transformed into zeta potential. Most colloidal particles have negative zeta potential values ranging from about -100 to -5 mV. Surface charges prevent the agglomeration of nanoparticles polymer dispersions because of strong electrostatic repulsion, thereby enhancing the stability of the nanoparticles. The zeta potential can also provide information regarding the nature of material encapsulated within the nanocapsule or coated onto the surface [67].
Analysis of structural changes of the free protein sample and protein nanoparticles is imperative to understand the nature of modifications taking place in the protein in terms of confirmation, folding, chemical bonding, etc., during the synthesis of nanoparticles.
X-ray diffraction: One of the techniques for this purpose is X-ray diffraction (XRD) which is the primary tool for investigating the structure of crystalline materials, from atomic arrangement to crystallite size and imperfections. XRD also analyzes the phase composition, crystallite size and shape, lattic distortions and faulting, composition variations, orientation and in situ structure development of the nanoparticles. Usually, the XRD pattern is obtained by illuminating the sample with an x-ray source (Copper Kα line) with wavelength of 1.54Å and scanning the diffraction within a certain range of the angle 2θ [68].
Fourier transform infrared spectroscopy: Another technique to supplement XRD is Fourier transform infrared spectroscopy (FTIR). The advantage of FTIR over crystallographic techniques is its capability to provide information about the structural details with greater spatial and temporal resolution [64]. The sample used for characterization is usually lyophilized nanoparticles in minute quantities. The basic principle that governs is that the bonds and groups of bonds vibrate at characteristic frequencies. A molecule that is exposed to infrared rays absorbs infrared energy at frequencies which are characteristic of that molecule. FTIR analysis is carried out by illuminating the sample with a modulated IR beam. The sample transmittance and reflectance of the infrared rays at different frequencies is translated into an IR absorption plot, which is then analyzed and matched with known signatures of identified materials in the FTIR library [69,70].
Chitosan NPs have numerous applications in pharmaceutical, biological and medical discipline.
Drug delivery to the lungs has various advantages such as rapid and sustained drug delivery, high efficacy, no first pass metabolism and both local and systemic effects can be achieved. The factors which contribute to the enhanced drug delivery via lungs are large surface area of lungs, thin absorption barrier and high vascularity [71]. Islam and Ferro in their review claimed that drug delivery to the lungs can also be achieved by chitosan-based NPs. The positive charge of the Chitosan provides it mucoadhesive properties which increases potential for drug absorption and this positive charge also open the tight junctions which lead to an increased uptake. Chitosan also possesses antibacterial properties which provide an additional benefit of fighting the pulmonary bacterial infections [72]. The effects of various drugs have been improved with the help of Chitosan. Using Chitosan as a polymer a nanoparticles dry powder inhalation of rifampicin, an anti-tubercular drug, was formulated. This formulation has shown sustained drug release until 24 hour and no toxic effects were shown to both the cell and organ [73,74]. Jafarinejad, et al. developed chitosan NPs for the pulmonary delivery of itraconazole (anti-fungal drug) as a dry powder formulation due to its low solubility when administered orally. This resulted in an increased pulmonary deposition of intraconazole. Petkar, et al. developed octanoyl chitosan NPs loaded with rifampicin for pulmonary delivery. This development improved sustained release and stability of rifampicin [75]. Debnath, et al. administered prothionamide (antitubercular drug) in the form of nanoparticle via pulmonary route and these NPs were coated with chitosan. This modification improved the residence time of the drug in the lungs [76].
Nasal delivery is a non-invasive technique through which, the delivered drugs reach the brain, the respiratory system and/or the systemic circulation. Chitosan increases the permeability of the hydrophilic drugs, nucleic acids, proteins and peptides across the nasal epithelium as they pose difficulty due to their low permeability [77,78].
Chitosan has been widely used for mucosal and systemic vaccine delivery and also in preparation of DNA mucosal vaccines. It is used in mucosal vaccine delivery as it promotes absorption which enhances mucosal immune response [79,80].
Chitosan has been widely used for mucosal and systemic vaccine delivery and also in preparation of DNA mucosal vaccines. It is used in mucosal vaccine delivery as it promotes absorption which enhances mucosal immune response [81]. Mainly, the vaccine administered through oral route requires the use of NPs. Nasal route vaccines are also there. In oral delivery vaccine the main target are Peyer's patches. Due to the nanoparticle system, the vaccine is protected from enzymatic degradation while going to the mucosal tissue and taken up by the M-cells. On the other hand, the nasal administered vaccines remain in the nasal cavity for about 15 minutes only; have to be transported over a very small distance and to be protected from low pH and enzymatic degradation [82-86]. Chitosan vaccines containing influenza, diphtheria and pertussis antigens for nasal delivery were prepared by Illum, et al. In systemic vaccine delivery, chitosan acts as an adjuvant. Activation of macrophages occurs after the uptake of chitosan [87-89]. Chitosan has been widely used for DNA mucosal vaccines. Illum, et al. developed a chitosan-based DNA flu vaccine. Incorporation of plasmid pCMVArah2 encoding peanut allergen gene into chitosan NPs showed good antigen expression and good protection after oral administration in mice. Vaccine [90,91].
On a laboratory scale, there are various options available for the preparation of PLGA nanoparticles by emulsion based technology platforms. Different nano-encapsulation methods lead to the formation of PLGA nanoparticles with different qualities. Key aspects discussed from the manufacturing perspective are size uniformity of nanoparticle populations, drug encapsulation efficiency, mixing device, solvent type, solvent residue in nanoparticles, and mixing device. Other foremost considerations that should be given to product development include scalability of a nanoencapsulation process, reproducibility, nanoparticle characterization, and quality control. Also considered as formidable hurdles are modulating nanoparticle functionality, programmability and target ability; adjusting drug release rate with minimal burst; and correlating in vitro - in vivo behavior of nanoparticles. Relevant issues have been well discussed elsewhere [92,93].
Nanoparticles are effective in drug delivery and also enhance the therapeutic efficacy of the drugs. Their positive charge makes them helpful in pulmonary drug delivery. They increase the permeability of various drugs making them available for nasal delivery. They act as an adjuvant in vaccine delivery. Overall, these applications of NPs are due to its physiochemical properties. These include biocompatibility, biodegradability, mucoadhesive character, absorption enhancing capability and in situ gelling property. However, a critical examination regarding the toxicity and safety issues of chitosan NPs and its manufacturing technique need to be investigated very closely.
Even though PLGA nanoparticles have already demonstrated great potentials as drug carriers, many overwhelming tasks still remain to be solved for their clinical translation. Before rushing into the investigation of in vivo behaviors of PLGA nanoparticles, it is imperative to make an in-depth assessment of technological aspects of a nanoencapsulation method of choice. It is also critical to carefully assess the quality of nanoparticles. In the practice of general emulsion-templated nanoencapsulation processes, mixing devices create zones of energy density in which high pressure, shear, turbulence, and/or cavitation drive the breakups of large emulsion droplets into nanosized ones. High-energy, high-shear, and high-pressure mixing devices damage the structural integrity of fragile macromolecules. Preparing PLGA nanoparticles with high loads of hydrophilic drugs often remains challenging. In addition, the mixing of a polymeric dispersed phase with antisolvent follows a bulk process, which lacks the ability to precisely direct the process of emulsification and the rate of PLGA precipitation. As a result, this procedure gives rise to the formation of a heterogeneous nanoparticle population with regard to mean diameter, size distribution, morphology, and drug content. Therefore, selecting or developing a nanoencapsulation method must surmount the aforementioned obstacles. At the same time, the nanoencapsulation method should be able to produce nanoparticles that meet the desired standards in a consistent manner.