Myeloproliferative neoplasms (MPNs) are clonal hematopoietic stem cell disorders with shared gene mutations, excess blood production and wide range of clinicopathological changes. JAK2, CALR and MPL gene mutation study has become one of the major criteria in the diagnosis of MPNs. This study focused on the common laboratory findings in patients with genetically proven MPNs at age 50 and younger with clinical correlation and follow up. There were 54 patients in this study, including 36 essential thrombocythemia (66.7%), 13 polycythemia vera (24.1%), 2 primary myelofibrosis (3.7%) and 3 MPN-unclassifiable (5.5%). The male/female ratio was 1/2 in patients with essential thrombocythemia, and about 1/1 in patients with other types of MPNs. Thrombocytosis and abnormal megakaryocytic proliferation were the most common findings in peripheral blood and bone marrow biopsy specimens, 88.9% and 96.8% respectively. Thrombotic event was recorded more often in patients with polycythemia vera than essential thrombocythemia, 30.1% and 19.4% respectively. Myelofibrosis, evolving acute myeloid leukemia and disease related mortality were seen only in patients with polycythemia vera, but not in patients with essential thrombocythemia. The findings in this study suggested that genetically induced abnormal megakaryocytic proliferation might play a critical role in the early pathogenesis of MPNs. With an increased risk for thrombotic event and disease progression, patients with polycythemia vera may need more follow up evaluation and therapeutic intervention than those with essential thrombocythemia. Anti-megakaryocytic therapy may be a future direction in the management of young MPN patients.
Myeloproliferative neoplasm, Essential thrombocythemia, Polycythemia vera, Primary myelofibrosis, Bone marrow biopsy, Thrombocytosis
MPNs: Myeloproliferative Neoplasms; ET: Essential Thrombocythemia; PV: Polycythemia Vera; PMF: Primary Myelofibrosis; CBC: Complete Blood Count; FISH: Fluorescence In Situ Hybridization; MDS: Myelodysplastic Syndrome; AML: Acute Myeloid Leukemia; PCR: Polymerase Chain Reaction; JAK2: Janus Kinase 2; CALR: Calreticulin; MPL: Myeloproliferative Leukemia Protein
Myeloproliferative neoplasms (MPNs) - essential thrombocythemia (ET), polycythemia vera (PV), and primary myelofibrosis (PMF) - are clonal hematopoietic stem cell disorders with shared gene mutations, excess blood production and wide range of clinicopathological changes [1,2]. Each type of MPNs is capable of evolving into another type and transforming into acute leukemia or bone marrow failure [3,4].
MPNs are more commonly seen in patients in their 60's or 70's [5,6]. Over 85% of patients with MPNs have a mutually exclusive mutation in one of the following three genes: JAK2, CALR or MPL [7,8]. Genetic predisposition has shown to increase disease susceptibility in certain populations, and the incidence of each type of MPNs is associated with some genetic variations including gender and age [9,10]. In recent years, testing for JAK2, CALR and MPL gene mutation has become a major criterion for the diagnosis of MPNs [11,12], and has facilitated the diagnosis of MPNs, especially in younger patients at an early stage of disease. This study is to evaluate the most common laboratory and pathological features in patients with genetically proven MPNs at age 50 and younger with clinical correlation and follow up.
The pathological reports and laboratory test results filed at the Department of Pathology, William Beaumont Hospital - Troy from 2006 to 2016 were reviewed. All patients were from the local population of a major US metropolitan in Midwest area. In total, 363 patients were diagnosed of MPNs with positive gene mutation studies for JAK2, CALR or MPL. Among them, 54 patients at age 50 and younger were selected for further analysis.
All patients had somatic gene mutation studies for JAK2, CALR and MPL. Genomic DNA was isolated from either peripheral blood or bone marrow aspirate specimen, followed by polymerase chain reaction (PCR) procedure using a primer pair specific or allele-specific probe. Mutation analysis of the amplified PCR product was performed by high resolution capillary electrophoresis. The result was reported as positive or negative for mutation in JAK2 gene (V617F), JAK2 exon 12, exon 9 of the CALR gene or 2 mutations of MPL gene (W515).
All patients had bone marrow biopsy samples and/or peripheral blood samples for routine laboratory and pathological evaluations. Bone marrow aspirate and peripheral blood smears were prepared with Giemsa and Wright stains for cytological evaluation. Iron stain was performed on all bone marrow aspirates for the evaluation of ring sideroblasts. Hematoxylin and Eosin (H&E) stained slides of bone marrow core biopsy was prepared for histological examination.
Fluorescence In Situ hybridization (FISH) for BCR/ABL gene rearrangement and conventional cytogenetic studies were performed on all bone marrow aspirate samples. In patients without bone marrow biopsy, PCR study was performed on peripheral blood samples for BCR/ABL gene rearrangement in addition to JAK2, CALR and MPL gene mutations. Myelodysplastic syndrome (MDS) FISH panel was performed in patients with anemia for the evaluation of myelodysplasia as clinically indicated.
Electronic medical charts of these patients were retrospectively reviewed. Laboratory results and clinical information were collected including complete blood count (CBC) results and trend, erythropoietin level, splenomegaly, thrombotic events, antithrombotic agents, other related treatment received and clinical follow up. The follow up period was 1 to 10 years after the initial diagnosis of MPNs and positive gene mutation study.
There were 54 patients in this study, consisting of 14.9% (54/363) of the total patients with MPNs diagnosed during the same time period at the referring institute. The patients included 22 males and 32 females, with a median age of 42 years (17 to 50). As summarized in Table 1, there were 36 ET patients, 13 PV, 2 PMF and 3 MPN-unclassifiable (MPN-U).
Table 1: Diagnosis of MPNs and related gene mutation study results. View Table 1
All patients were positive for one of the gene mutations. JAK2, CALR and MPL gene mutation appeared mutually exclusive and no double positivity was seen in any patient. When a bone marrow biopsy was available for examination, the diagnosis of MPNs was made according to 2004 and 2008 WHO classification system [2,13]. When no bone marrow biopsy was available, the diagnosis of MPNs was made with combination of WHO classification criteria, laboratory test results and the clinical findings such as splenomegaly and thrombotic events.
The common hematology laboratory findings were shown in Figure 1. Fifty-one (94.4%) patients presented with abnormal CBC findings. Thrombocytosis (88.9%) was most common followed by erythrocytosis (25.9%), anemia (22.2%) and leukocytosis (11.1%). No thrombocytopenia or leukopenia was recorded in any patient.
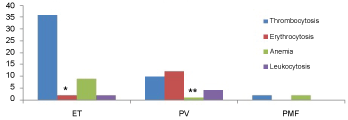 Figure 1: Common CBC findings in patients with MPNs.
Figure 1: Common CBC findings in patients with MPNs.
*2 patients with ET presenting with mild erythrocytosis below the limit required for the diagnosis of PV.
**1 patient with PV and post-polycythemic myelofibrosis presenting with anemia. View Figure 1
The 36 ET patients had a male/female ratio of 12/24 (Table 2). Among them, JAK2 gene mutation was positive in 29 patients with a male/female ratio of 9/20. CALR gene mutation was positive in 3 male and 3 female patients respectively. MPL gene mutation was positive in 1 female patient.
Table 2: Male and female ratio in patients with MPNs at age 50 and younger. View Table 2
All patients presented with persistent thrombocytosis. Two patients also had mild erythrocytosis. Since the erythrocytosis remained below the limits for the diagnosis of PV as recommended by WHO criteria with normal erythropoietin level, the diagnosis of ET maintained. Anemia was seen in 9 patients and mild leukocytosis in 2 patients.
The 13 PV patients had a nearly even male/female ratio (7/6). Twelve patients had JAK2 (V617F) gene mutation and 1 had JAK2 exon 12 gene mutation. All patients had significant erythrocytosis. Two patients initially presented with thrombocytosis only. However, significant erythrocytosis developed 10 and 13 months later respectively during the follow up period supported the diagnosis of PV.
Two patients showed myelofibrosis in bone marrow biopsy evaluation with clinical and pathological findings consistent with post-polycythemic myelofibrosis. Another patient was initially diagnosed of PV based on clinical and peripheral blood findings and 6 years later the bone marrow biopsy showed evolving acute myeloid leukemia (AML) with abnormal cytogenetic findings (see below).
PMF was diagnosis in 2 patients, 1 male and 1 female, both with CALR gene mutation. Both patients presented with thrombocytosis and anemia and had bone marrow biopsies available for histological confirmation.
There were 2 patients positive for JAK2 and 1 patient positive for CALR gene mutation showing no abnormal CBC findings during the follow up period and having no bone marrow biopsy for histological examination. Gene mutation studies in these patients were promoted by clinical considerations and the diagnosis of MPN-U was made based on combined clinical and genetic findings.
Bone marrow biopsies were available for histological examination in 31 patients, including 19 ET, 10 PV and 2 PMF (53%, 77% and 100% respectively). The common bone marrow histological findings were shown in Figure 2. Megakaryocytic proliferation and atypia were most common (96.8%). In addition to hypercellularity, panmyelosis and erythroid hyperplasia were also more common in patients with PV. Iron stain was performed on all bone marrow aspirates and no ring sideroblasts were identified in any patient.
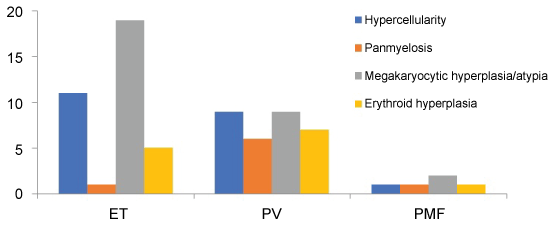 Figure 2: Common bone marrow pathological findings in patents with MPNs. View Figure 2
Figure 2: Common bone marrow pathological findings in patents with MPNs. View Figure 2
BCR/ABL gene rearrangement status was evaluated by FISH on bone marrow aspirate and/or by PCR on peripheral blood. No BCR/ABL gene rearrangement was identified in any patient. MDS FISH panel was performed on the bone marrow aspirates from patients with anemia and yield no abnormal findings.
Thirty-one patients had bone marrow aspirates available for conventional cytogenetic analysis. One PV patient with evolving AML showed abnormal karyotype: 47,XY,+del(1)(p13),der(20)t(1;20)(p13;q12). Studies revealed normal karyotype in the rest 30 patients.
Thrombotic events were recorded in 11 patients; 7 with ET and 4 with PV (19.4% and 30.8% respectively). Thirty-three patients with the diagnosis of MPNs in this study received antithrombotic agents, including 21 ET patients, 8 PV, 2 PMF and 2 MPN-U. The most common medication was aspirin, recorded in 31 patients. The other medications were much less common and generally used in combination with aspirin: lovenox and warfarin in 4 and 2 patients with ET respectively; apixaban and plavix each in a patient with PV.
In addition to anti-thrombotic agents, 11 patients received hydroxyurea (6 ET, 4 PV and 1 MPN-U). The patient with evolving AML was treated with chemotherapy and bone marrow transplantation and was in remission for more than 5 years in follow up. Another patient with post-polycythemic myelofibrosis died during the follow up due to bone marrow failure and multiple complications. No other mortality was recorded in the patients studied.
In this study of patients at age 50 and younger with genetically proven MPNs, ET was the most common subtype with a female predominance, similar to the previously reported data before gene mutation studies became available [5,14]. There was no apparent gender difference observed in patients with PV and PMF, although the number of patients was limited in each group.
Thrombocytosis was the most common CBC finding in these patients. In addition to thrombocytosis, anemia was also common in patients with ET and was the clinical indication for pathological evaluation of myelodysplasia. Mild erythrocytosis may be present occasionally in patients with ET. Follow up CBC evaluation with trend and erythropoietin level will help to distinguish these patients from PV. Histological examination of bone marrow biopsy showed consistent megakaryocytic proliferation and atypia. Most patients had hypercellular bone marrow. Panmyelosis and erythroid hyperplasia, however, were more common in patients with PV.
MPNs are genetically and phenotypically related diseases [15-17]. Occasionally, patients may present with thrombocytosis as the only abnormal laboratory finding at the initial evaluation, which makes the diagnosis of MPN more consistent with ET. However, follow up laboratory evaluation may yield additional findings which could alter the diagnosis. Two patients in this study were initially diagnosed as MPN, most consistent with ET based on the findings of positive JAK2 gene mutation and thrombocytosis in peripheral blood. However, follow up study showed persistent erythrocytosis and other findings fulfilling the diagnosis of PV.
Patients with different subtype of MPNs may show distinct clinical and laboratory features, possible related to the patient's gender and age [18,19]. In this study, patients with ET appeared to have a relatively stable clinical course with lower thrombotic risk, no myelofibrosis or evolving acute leukemia, and no disease related mortality reported. On the other hand, patients with PV had higher risk of thrombosis. One PV patient showed progression to evolving AML with abnormal karyotype and another patient had post-polycythemic myelofibrosis with bone marrow failure and mortality.
Thrombosis is a significant complication in patients with MPNs and anti-thrombotic therapy is a major component of patient management [20-22]. In this study, 30.8% patients with PV and 19.4% patients with ET had thrombotic episodes. Thirty-three patients received antithrombotic therapy. Aspirin was the most common anti-thrombotic agent prescribed in these patients, plus lovenox and warfarin in a few patients. Apixaban and plavix were also used occasionally. In addition to anti-thrombotic agents, 11 patients were also treated with hydroxyurea as recommended by the clinical guidelines, although the efficacy of hydroxyurea therapy in these patients has been controversial [23-25].
In this study, ET appears the most common subtype of genetically proven MPNs in patients at age 50 and younger with a female predominance. Thrombocytosis and abnormal megakaryocytic proliferation were the most common laboratory findings in peripheral blood and bone marrow suggested that genetically induced abnormal megakaryocytic proliferation may play a critical role in the development of MPNs, especially at the early stage of disease. Anti-megakaryocytic therapy may be a future direction in the management of patients with MPNs at a younger age. Patients with ET had increased risk for thrombosis, but in overall a relatively stable clinical course. Patients with PV, on the other hand, had higher risk for thrombosis and disease progression including evolving AML and myelofibrosis with bone marrow failure and mortality. We acknowledge that this was a retrospective study on a small cohort of younger patients with MPNs. Some patients had limited clinical information available for analysis. Further studies with more patients and longer clinical follow up are needed to investigate the early pathogenesis of MPNs.
Department of Pathology, William Beaumont Hospital, Troy, Michigan.