Congenital pulmonary airway malformation or CPAM is a rare anomaly which affects specifically one and usually lower lung lobe. In most cases it is detected during prenatal life with foetal ultrasound, but it`s not uncommon to find CPAM in background of frequent respiratory infections in childhood, rarely even in adulthood. In this paper, our aim is to present an atypical presentation of CPAM type I which we had difficulties to diagnose. Our case shows CPAM as a substrate for development of tension pneumothorax in 5-months old infant facilitated by acute bronchitis. After initial thoracic drainage and regression of pneumothorax a recurrent pneumothorax was developed and consequently CPAM was suspected. Following CT scans showed CPAM lesion in lower right lung lobe with mediastinal shift and consolidation of adjacent lung tissue. Parenchyma sparring lung resection was done and histological analysis verified CPAM type I lesion. No postoperative complications occurred and infant`s further development was normal during regular follow-up.
Congenital pulmonary airway malformation, Pneumothorax, Acute bronchitis
Congenital malformation of the airways or CPAM is a sporadic disease and represents a dysplastic developmental malformation of the lungs. It occurs with an incidence of 1: 25,000 to 1: 35,000 live births, but many authors suggest that its incidence is underestimated and that hypothetically thousands of people with this malformation live their entire lives without complications [1-5]. CPAM accounts for about 25-30% of all congenital lung malformations, and about 70% of cases is diagnosed in utero [1,6,7]. The rest is diagnosed postnatally after the development of respiratory symptomatology or incidentally by X-rays or CT scans of the thoracic organs. The main characteristics of the malformation are a direct connection with the tracheobronchial tree and vascularization through the pulmonary circulation. CPAM most often involves one lung lobe, but atypical forms may involve multiple lobes, the entire lung on one side or even whole lungs bilaterally [6-8]. The middle lung lobes are the least, and the lower lobes are the most commonly affected [7,8]. The malformation was divided by Stocker into five types, and type I is the most common type of CPAM. In type I there is one or more cysts with a diameter of 2-10 cm with one dominant cyst and often mediastinal displacement. The clinical course of the disease is variable and includes asymptomatic presentation up to life-threatening conditions such as fetal hydrops or respiratory failure in the newborn. Treatment includes methods of fetal surgery or postnatal lung resections the timing of which depends on the age of child and the existing symptoms of the disease.
A 5-month-old male infant was administered in the emergency pediatric ambulance with clinical signs of acute bronchitis. He had coughed for three days and the night before administration to the emergency room he became dyspneic. Earlier that day, in primary care, he was treated with salbutamol and corticosteroid therapy. This was his first respiratory infection. In antenatal period there was no detection of mediastinal shift on fetal ultrasound, nor was there a suspicion of a congenital lung lesion. Initial X-rays showed right-sided tension pneumothorax with left mediastinal displacement (Figure 1a). Emergency thoracic drainage was indicated and after drainage the child was transferred to the ICU. Respiratory status of infant improved and on control X-rays we monitored regression of pneumothorax without signs of cystic formations or changes in the lung parenchyma (Figure 1b). The drain was removed on the 5th postoperative day. However, milder respiratory symptomatology developed again after 3 days and recurrent pneumothorax with mediastinal shift was found on the repeated X-rays (Figure 1c) which led to a second thoracic tube placement. Because of the suspicion of a congenital lung lesion, CT diagnosis was indicated. On CT scans, a cystic malformation of the lateral segment of the lower right lung lobe was found with atypical consolidated lung tissue at the border with the middle lobe and herniation of the anterior lobe. There was also residual pneumothorax with left mediastinal displacement (Figure 1d). Due to a highly suspected congenital airway malformation found on CT scans, surgery was indicated. Preoperatively we performed bronchoscopy and showed membrane with one-way valvular mechanism at the entrance in lateral segment of inferior right lung lobe which explains tension pneumothorax (Figure 2). After that, a right thoracotomy was performed and one large cystic formation (3.5 × 4 × 1cm) of the right posterior lower lobe of the lung was found with 2 more smaller cysts about 1.5 cm in diameter (Figure 3). The other parts of the lower lobe were intraoperatively normally formed. Atypical resection was made to the healthy edges of the lungs, and the remaining part of the lower lobe was re-expanded. The infant was administered in the ICU where regular postoperative recovery with no complications followed. Histological analysis confirmed CPAM type I (Figure 4). On outpatients controls child was asymptomatic and thriving normally. Postoperative X-rays showed normal lung expansion (Figure 5).
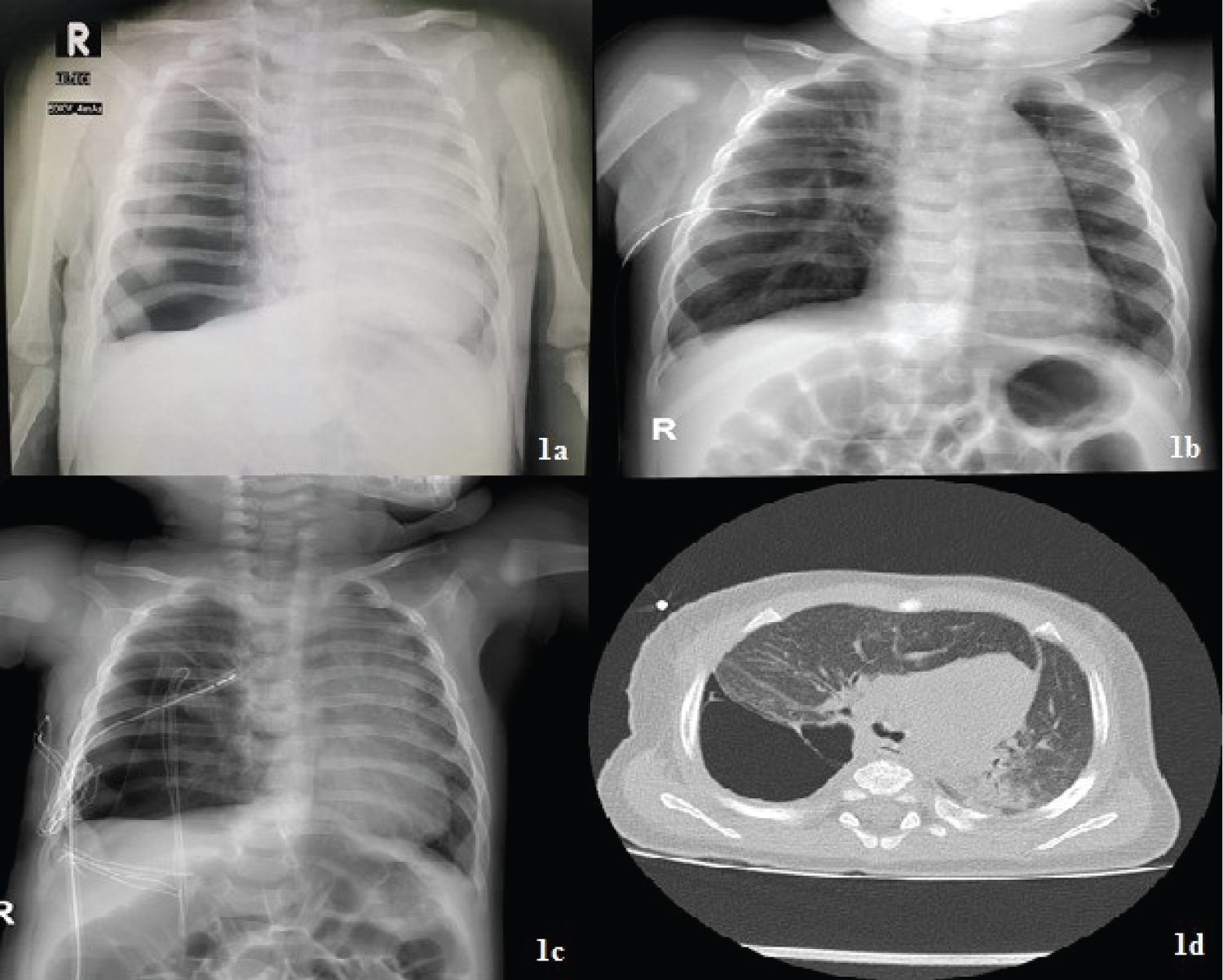 Figure 1: a) right-sided pneumothorax with large left-sided mediastinal shift; b) X-rays after initial thoracic drainage shows no signs of CPAM; c) X-rays showing 17.5 mm recurent pneumothorax with large mediastinal shift and chest tube in situ; d) CT scan shows cystic malformation of right lower lung lobe with mediastinal shift and consolidation of adjacent parenchyma.
View Figure 1
Figure 1: a) right-sided pneumothorax with large left-sided mediastinal shift; b) X-rays after initial thoracic drainage shows no signs of CPAM; c) X-rays showing 17.5 mm recurent pneumothorax with large mediastinal shift and chest tube in situ; d) CT scan shows cystic malformation of right lower lung lobe with mediastinal shift and consolidation of adjacent parenchyma.
View Figure 1
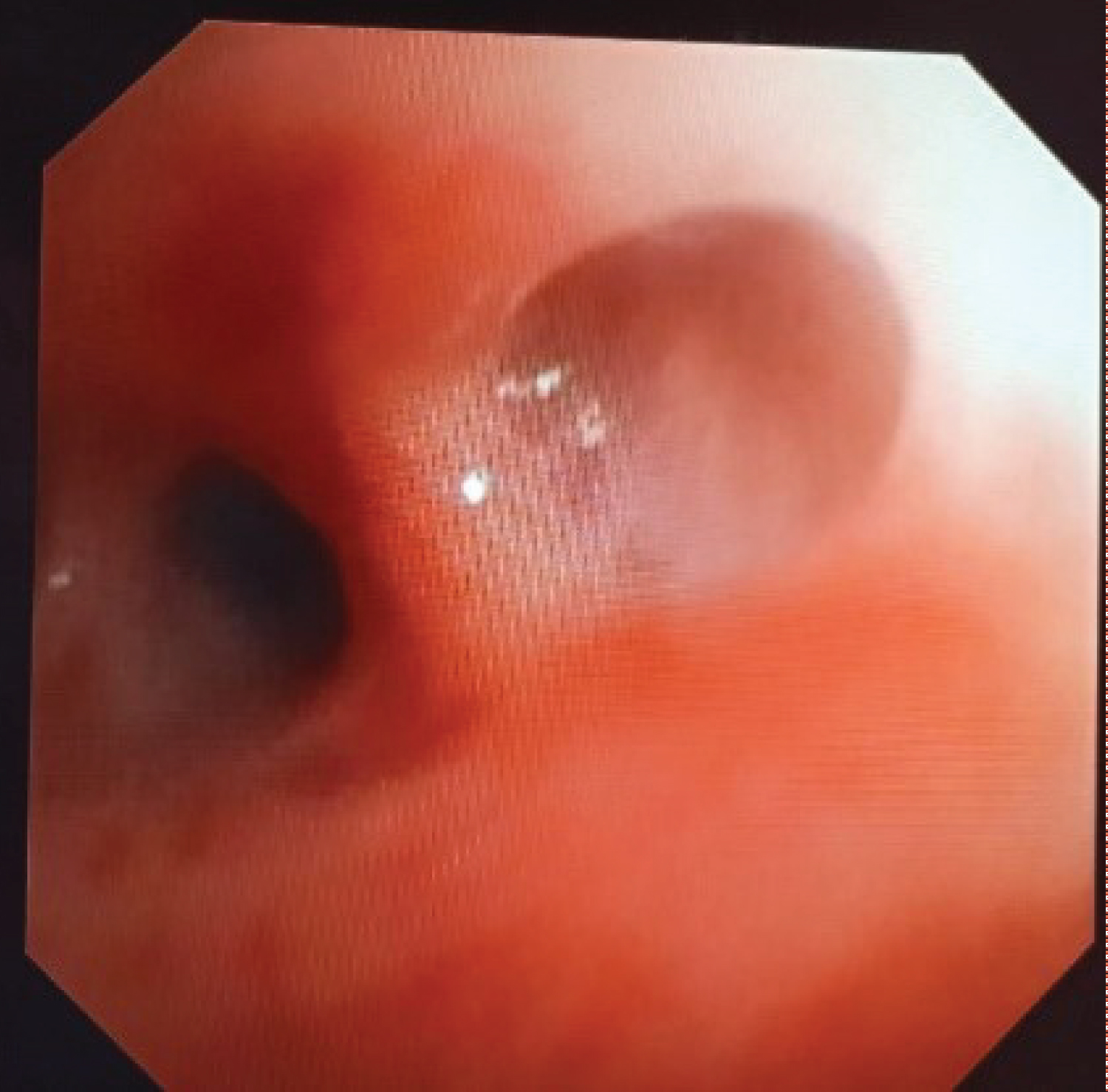 Figure 2: Preoperative bronchoscopy showing membrane at the entrance in lateral segment of lower right lung lobe.
View Figure 2
Figure 2: Preoperative bronchoscopy showing membrane at the entrance in lateral segment of lower right lung lobe.
View Figure 2
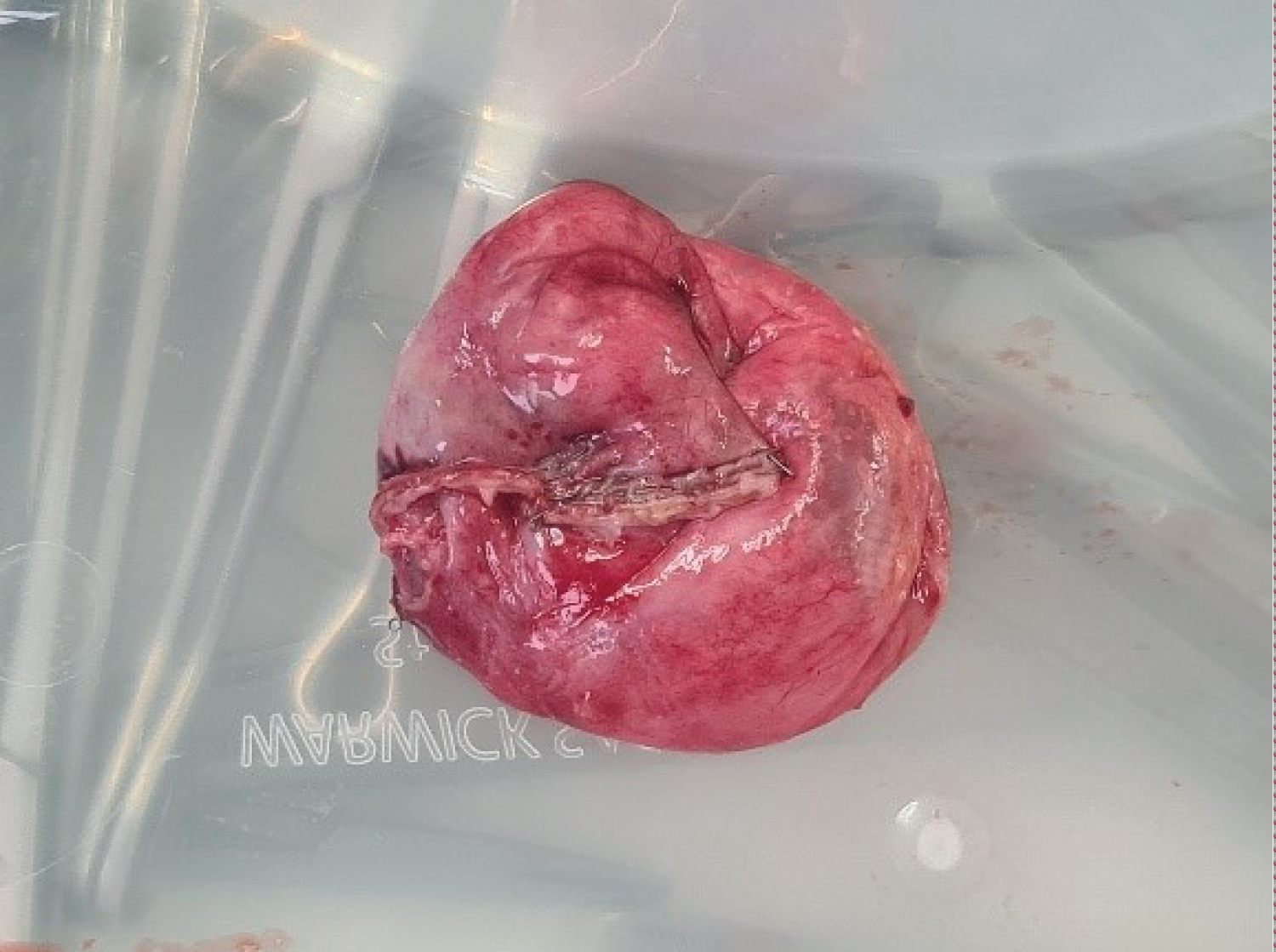 Figure 3: Resected malformation.
View Figure 3
Figure 3: Resected malformation.
View Figure 3
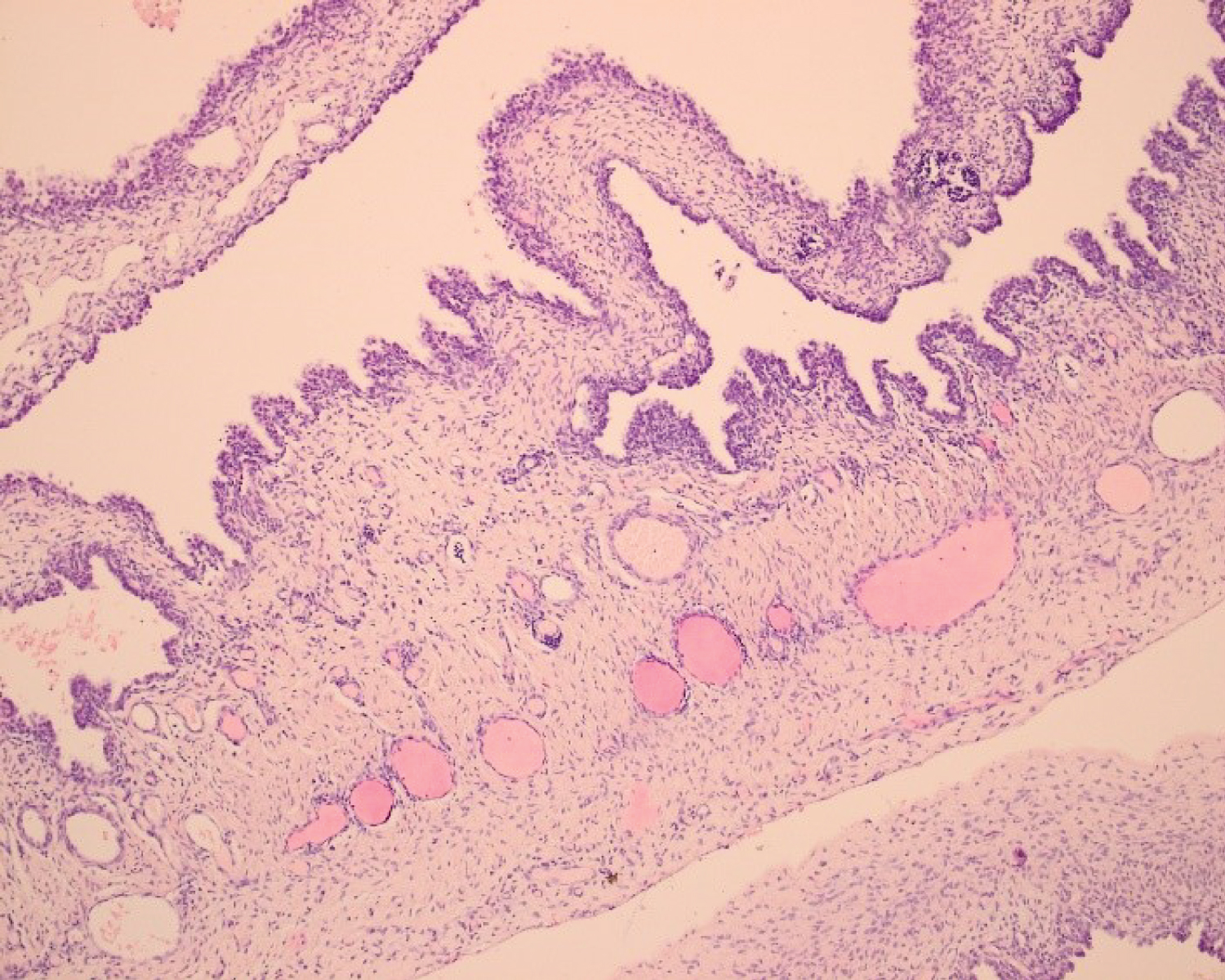 Figure 4: Variably sized cysts lined by pseudostratified columnar-type and flattened epithelium, HEx100.
View Figure 4
Figure 4: Variably sized cysts lined by pseudostratified columnar-type and flattened epithelium, HEx100.
View Figure 4
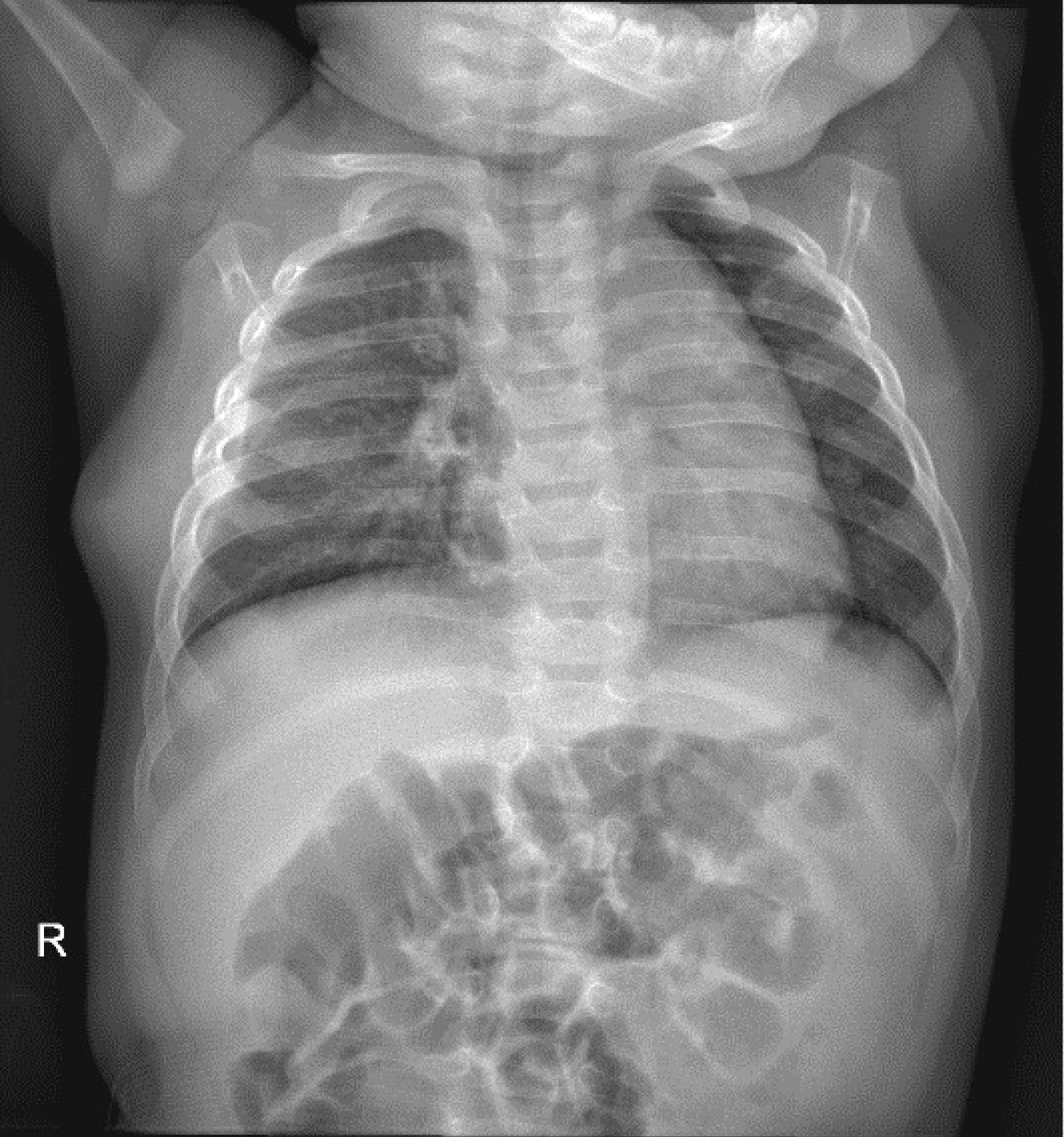 Figure 5: Postoperative X-rays (1 month after surgery).
View Figure 5
Figure 5: Postoperative X-rays (1 month after surgery).
View Figure 5
CPAM is divided by Stocker into five histological types. Tip I and IV are characterized with large cysts so it can be very difficult to distinguish them on antenatal ultrasound and even on postnatal CT scans, especially if a child develops pneumothorax or respiratory infection. Children with CPAM are born mostly as full-term neonates of regular birth weight [9]. Most babies are born asymptomatic, but about 30% of neonates with CPAM are at risk of acute respiratory failure at birth or show signs of respiratory distress in the neonatal period [10,11]. Older symptomatic children usually present with frequent respiratory infections during the first years of life [11,12]. In children with CPAM who have not been operated at the neonatal age, the risk of infection in the first year of life is 10-30%, and the first symptoms in primarily asymptomatic children appear most often by 7 months of age [13-16]. In our case, the child was asymptomatic in the neonatal period and presented with acute bronchitis complicated by pneumothorax at 5 months of age.
The standard diagnostic tool for CPAM is antenatal ultrasound which detects about 70% of CPAM cases. The malformation is detectable in the second trimester of pregnancy and is most often verified during an anomaly scan [6]. Since ultrasound has overlapping criteria for CPAM and other lung malformations, all formations are prenatally referred as congenital lung lesions. In case of unclear diagnosis, fetal MRI can be performed. In our patient, there was no suspected congenital lung malformation, the fetus developed normally and the pregnancy and childbirth went without complications.
Postnatal diagnostic methods include X-rays and CT scans. In any case, recurrent pneumothorax or altered non-regressive lung parenchyma are an indication for CT of the thorax. Postnatal X-rays are insufficient to assess CPAM which will be seen as a soft tissue shadow with possible fluid levels due to slow fluid clearance within the formation or it will not be detectable like in our case [7]. Computed tomography (CT) is standard diagnostic method for these malformations, since MRI is insufficient in the assessment of intra-parenchymal lung lesions [17]. CPAM is most often seen as a multi-cystic formation filled with air with thin septa, and types I, II, and III can be distinguished with certainty [11,18]. Considering our case, there were no signs of CPAM on postnatal X-rays, but CT showed typical characteristics of macrocystic CPAM although it did not clearly differentiate whether it is type I or IV because it was mimicked with pneumothorax.
Surgical treatment of CPAM is very complex and includes procedures in the field of fetal, neonatal and pediatric surgery. In symptomatic infants and children, it is absolutely necessary to do X-rays and early CT of the thoracic organs. After radiological confirmation, early surgery is planned. Lobectomy is the option of choice in the case of unilobular presentation. But atypical parenchyma sparring resections can spare the child postoperative mechanical ventilation. Also, recent research has focused on the efficacy of non-anatomical lung resections due to the fact that fragments of residual lung tissue have a high capacity for expansion and functional recovery by age 9 [19]. If lung resection is performed at an earlier age, like in our case, there is more room for compensatory growth of a healthy remaining lung. However, recent work suggests comparatively equal lung function in children who had early surgery and those who underwent surgery at a later age [20]. It is important to note, although, that in the case of symptoms or infection at the time of the procedure, lung function may be reduced in the future [21].
CPAM is a rare congenital malformation with wide spectrum of clinical presentation. In any case of pneumothorax in neonate or infant a suspicion for CPAM must be out ruled. Diagnostic algorithm with obligatory CT scans and early surgery has to be performed if a child develops complications such as infection or pneumothorax to prevent further damaging of parenchyma and provide a chance compensatory lung growth. Also, it has been proven that later in life, CPAM (especially type I and IV) are connected with development of malignant lung diseases such as bronchioalveolar carcinoma or pleuro-pulmonal blastoma. Regarding type of resection, lobectomy was always a method of choice, but recent work suggests that atypical resections can provide better functional recovery of the lungs.