Cystic Fibrosis (CF) is a frequent recessive autosomal disease characterized by a wide variety of sequence alterations in the Cystic Fibrosis Transmembrane Regulator (CFTR) gene [1,2]. Since the initial characterisation of the CFTR gene in 1989 [1], more than 2000 mutations have been reported to the Cystic Fibrosis Consortium (http://www.genet.sickkids.on.ca/cftr). These mutations can alter CFTR function by mechanisms that range from the loss of chloride conductance to complete absence of protein synthesis. In fact according to the impact of the genetic defection, CFTR mutations could be grouped in six classes: Class I induces protein synthesis defect due to premature Stop codon or alteration of critical RNA signal results in failure to synthesize full-length CFTR, class II induces folding or trafficking defect, class III and IV affect the CFTR function at the cell surface respectively the gating or the conductance, Class V and VI induce a diminution of the quantity of functional CFTR respectively by splicing defect or by CFTR stability decrease [3,4].
The history of Lebanon, characterized by flows of different ethnic groups, has enabled the introduction of new genes and a wide variety of genetic diseases. The early development of medical services in Lebanon has facilitated the detection of many inherent disorders. CF figures among the 184 reported genetic diseases of the Lebanese population. Even though no epidemiological studies were performed to determine the CF incidence in Lebanon, multiple factors advocate that it could be relatively high.
1) The Lebanese CFTR mutational spectrum [5,6] uncovered the major European and minor Arab influences on the Lebanese CFTR gene pool.
2) The lack of intra-communities mixing [7] and the consanguineous marriage that has a prevalence of 35.5% especially in the non-Christian communities and suburbs of Beirut [8] tend to extent the incidence of CF in Lebanon. Consanguinity that had historically a higher rate is an essential cause of new CF Lebanese patients. 50% of CF child in Lebanon were issued from a consanguineous marriage [5,6].
3) The enumerated CF cases in Arabs along the last two decades had revealed a decreasing incidence while ascending from the Mediterranean Sea to Arab Peninsula. An extrapolation to these observations presumes a high incidence in Lebanon regarding to its geographical position.
In the Lebanese population, the incidence of both common and rare genetic diseases is relatively high compared to neighbor countries [5] due to the existence of several communities and consanguineous marriage. Around 17% of the patients admitted to the Pediatric Service of American University in Beirut during 1961, 1966 and 1971, were found to suffer from a genetically caused or predisposed disorder [9]. Even thought the first Arab CF child was detected in Lebanon in 1958 [10], few accurate data were presented during the following years to study this disease in the Lebanese population.
However, the mutational CFTR spectrum of the Lebanese population was roughly elucidated in two previous studies [5,6]. The major characteristic of this spectrum concerns the p. Asn1303Lys (N1303K) mutation. This widespread mutation appears to have the highest worldwide frequency in Lebanon. Soon after its identification, this mutation was characterized by its severity on the pancreas and the variability of the pulmonary status [11]. Despite the low effective in the previous studies, almost all N1303K Lebanese CF patients (homozygous or heterozygous carrying another severe mutation in trans) showed severe pancreatic and pulmonary phenotypes [5,6]. The presence of a complex allele may aggravate its clinical outcome [12,13]. Indeed, mutation effects can vary by virtue of association in cis with other mutations (complex allele), modifying phenotype severity, and can explain the variability of the CF phenotype in CF N1303K patients. More recently, splicing studies [14] showed in cellulo no impact of this mutation on the exon 24 skipping whereas the associated complex allele induces minor exon 7 skipping, suggesting that this complex allele c. [744-33GATT [6]; 869 + 11C > T, 3909C > G] may not affect the phenotype. So, the few reduced quantity of functional CFTR resulting from this complex allele is not sufficient to explain the phenotype severity. More interestingly, this substitution occurs in the nucleotide Binding domain 2 (NBD2) that initially was predicted to have an unessential role in a proper folding and plasma membrane trafficking [15], unlike the four other CFTR domains. However, despite this fact, N1303K is classified as a class II mutation that retains CFTR in the endoplasmic reticulum. After transient transfections of HeLa cells by plasmids pTCF WT or N1303K, coverslips were incubated with human anti-CFTR MAB25031 (Invitrogen) (1/400 in 1x PBS) for 1 h at 4 ℃ and after washing with 1x PBS, cells were incubated in the dark with the secondary antibody, conjugated to Alexa Fluor® 555 (Invitrogen) (1/800 in 1x PBS) for 40 min. Cells were washed three times with 1x PBS, and coverslips were mounted with Mowiol (Sigma) [16]. Images were obtained using a confocal microscope (Andor Revolution) equipped with a multi-line Argon laser (457 nm, 488 nm, 515 nm; total 30 mW) to visualise GFP, and with a HeNe-Green laser (543 nm; total 1.5 mW) to visualise CFTR. Thus, the N1303K-CFTR is incompletely glycosylated, presenting a disruption in its global conformation [17] and its gating activity [18,19]. This structural defect created by N1303K seems to be different from the one induced by p. Phe508del (F508del). Indeed, F508del-CFTR protein is corrected by VX-809 (Vertex) whereas this drug is inefficient at rescuing N1303K-CFTR [16]. VX-809 represents a class of CFTR corrector that specifically addresses the underlying processing defect in F508del-CFTR [20].
The CFTR mutational spectrum of the Lebanese population was obtained by the studies conducted on 36 alleles in 1997 [5] then on 44 alleles in 2000 [6]. The c.1521_1523delCTT (F508del), c.3909C > G (N1303K) and c.3846G > A (W1282X) are account for almost two third of the alleles, in both studies. The N1303K mutation is ranked in the second position (detected in 27% of the 44 alleles) or in the third position (detected in 9.4% of the 36 alleles). This mutation that is very common in the Mediterranean countries [21-26] seems to have the highest frequency in the Lebanese population.
Interestingly, the countries and geographic locations that have a high N1303K frequency (Tunisia, Algeria, South Spain, South France, and South Italy) were old Phoenician settled colonies or trading posts (Figure 1). Phoenician migration flows from current Lebanon, during the first millennium BCE, may have introduced new genes in these costal Mediterranean cities. Genetic Phoenician markers described elsewhere [27], could be typed in worldwide CF-alleles carrying N1303K to support this hypothesis. Moreover, for tracing the origin of the most common CFTR mutations (F508del, G542X and N1303K), several CFTR genetic markers have been used [28-31]. The haplotype at CFTR markers IVS1CA-J44-GATT-T854-TUB20 was always 21-1-6-1- 2 when present with the N1303K mutation as well for the c.1521_1523delCTT and c.1624G > T mutations [30,31]. This associated haplotype (GATT6), which has been also detected in our study, is only present in 1.4% of normal chromosome in actual European [30]. Thus, it has been postulated that these mutations arose from a none-European population [29]. Moreover, no population has presented this haplotype in a high enough frequency to be considered at the origin of the three most common mutations. It is also important to note here, that the highest frequency, even not sufficient, was detected in the Druze population of Palestine [30]. This small community is also settled in Lebanon.
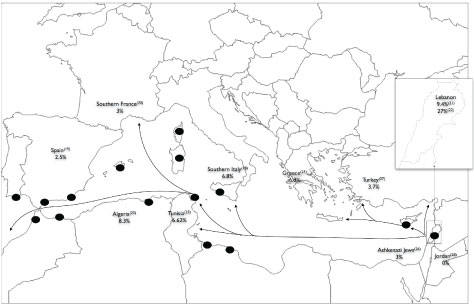 Figure 1: Frequencies of the N1303K mutation in Mediterranean countries and Phoenician migration.
Figure 1: Frequencies of the N1303K mutation in Mediterranean countries and Phoenician migration.
The black spots represent the most important Phoenician cities of the Mediterranean region. These cities were Phoenician colonies or trade centres. The arrows are major Phoenician migration and trade routes [5,6,22,25,26,32-41].
View Figure 1
Thus, it could consider that the origin of the N1303K mutation could be Phoenician, but no previous studies have been done in Lebanon (recent Phoenicia) to determine the frequency of the associated haplotype in normal CFTR chromosomes. Therefore, DNA samples should be sequenced from none-CF Lebanese at the CFTR-genetic markers. This added to sequencing of N1303K alleles in different populations at the Phoenician markers can provide conclusive evidence on the origin or not of the N1303K mutation.
This work was supported by Poitiers Hospital and University of Poitiers, France and ABCF2 mucoviscidose, France.
This work was supported by "association mucoviscidose: ABCF2". Raed Farhat received a fellowship from CNRS-Lebanon.