This study delves into the effects of secondary metabolites in Clerodendrum volubile on CYP2e1, a protein target implicated in hepatotoxicity. Among implicated in cis,cis-linoleic acid, 3-hydroxydecanoic acid, n-pentadecanoic acid, palmitic acid, and 3,5-dihydroxy-6-methyl-2,3-dihydro-4H-pyran-4-one demonstrate strong binding affinities and stability within the protein's active site compared to standard compounds with lower values. These compounds exhibited notable binding affinities against CYP2E1 with energies of -10.746, -10.203, -9.974, -9.177, and -8.447 kcal/mol, respectively. The inhibition of cytochromes P450 (CYPs) activities, responsible for procarcinogen activation, is implicated in the mechanism. This investigation focuses on the inhibitory potential of C. volubile compounds on CYP2E1 enzymatic activities to mitigate liver toxicity. Further analysis of the five hit compounds for drug-likeness and toxicity using SWISSADME reveals that all compounds possess desirable drug-like properties and are readily absorbable. The hit ligands effortlessly met the criteria molecular weight (MW) of ≤ 500 and violated none in Lipinski rule of 5 (Ro5).
Clerodendrum volubile , Phytochemicals, CYP2e1, Hepatoprotection, Pharmacokinetics
Hepatotoxicity is a significant health concern, and it is characterized by liver damage a life-threatening disease that causes hepatocyte death and inflammation of the liver. There are several causes of this clinical complication some of which are due to exposure to viral infection, improper drug use, excessive alcohol intake, and sepsis. Liver failure, is a severe consequence of abrupt massive hepatocyte death, evolving over days or weeks to a lethal outcome, with a clinical mortality of 25-75%. The liver is one of the cogent organs in the body, It is a major public health concern because the liver plays a crucial role in metabolism, detoxification, and immune function coupled with its functions in protein, lipid, and glucose metabolism, and synthesizes most plasma protein. Different injuries to liver cells result in a consistent sequence of events characterized by a sudden increase in aminotransferases, changes in mental function, and disruptions in blood clotting. The causes of acute liver failure encompass acetaminophen toxicity, hepatic ischemia, viral and autoimmune hepatitis, and drug-induced liver injury resulting from prescription medications as well as herbal and dietary supplements. Hepatotoxicity is a liver injury caused by various factors, including drugs, toxins, and viral infections. CYP2E1 protein is a transcription factor that plays a crucial role in the regulation of inflammatory responses, cell proliferation, and apoptosis. Cytochrome P450 2E1 (CYP2E1, EC 1.14. 13. n7) is a member of the cytochrome P450 mixed-function oxidase system, which is involved in the metabolism of xenobiotics in the body [1-3]. CYP2E1 has major contribution in liver diseases such as nonalcoholic fatty liver disease, alcoholic liver disease, and nonalcoholic steatohepatitis, probably through hepatic lipid peroxidation. The deleterious effects of CYP2E1 are also due to the fact that significant levels are located within mitochondria [4-6]. Chronic ethanol use can induce CYP2E1 activity, leading to a greater percentage of acetaminophen metabolized to NAPQI, increasing the risk of hepatotoxicity from acetaminophen use. Many scientists question the existence of an important ethanol-acetaminophen interaction [7,8]. Several studies have linked CYP2E1 activation to the pathogenesis of liver diseases, including hepatotoxicity.
CYP2E1 is considered one of the common pathogenic factors for both Alcoholic fatty liver diseases (AFLD) and non-alcoholic fatty liver diseases (NAFLD) due to its inducibility under these conditions with the increased ROS, causing deleterious effects on mitochondrial dysfunction and hepatotoxicities [4]. Therefore, the inhibition of CYP2E1 activation has been suggested as a potential therapeutic target for liver diseases [9]. CYP2E1 is associated with liver toxicity through two mechanisms: Firstly, by activating substrates into reactive metabolites, and secondly, by potentially generating reactive oxygen species (ROS). The lack of close coordination between substrate binding, oxygen binding, and electron delivery during catalysis can lead to the conversion of diatomic oxygen into superoxide or hydrogen peroxide, which is released instead of being utilized to monooxygenate the substrate [10]. These ROS have been implicated in various damaging events, including lipid peroxidation, protein oxidation, and DNA oxidation. Notably, CYP2E1 exhibits a significantly higher propensity for ROS production compared to other cytochrome P-450 enzymes, and some drugs have been explored for their potential in treating hepatotoxicity targeting the CYP2E1 protein. Several drugs have been used in the treatment of liver diseases, including Prednisolone. However, the adverse effects associated with these drugs have limited their use in clinical practice. Natural products have gained increasing attention in the treatment of liver diseases due to their low toxicity and potential therapeutic effects [11,12]. Flavonoids are a class of natural compounds that have been reported to protect against hepatotoxicity by inhibiting the CYP2E1 pathway [13]. C . volubile is a leafy vegetable found in certain regions of Nigeria, is not fully utilized despite its growing popularity due to its various benefits, such as its traditional uses, nutritional content, and potential therapeutic properties. Nonetheless, there is still much to uncover regarding the plant's pharmacological potential [14].
Molecular docking, however, offers a faster and more efficient way to screen for potential compounds with the desired pharmacological activity. By predicting the binding affinity of small molecules to target proteins, molecular docking enables researchers to identify compounds that are likely to bind to the target protein and exhibit the desired pharmacological activity [15]. This study aimed at investigating the molecular docking of C . volubile compounds on hepatotoxicity targeting CYP2E1 protein with resveratrol as a natural inhibitor, Sulforaphane, and chlormethiazole drugs are also used as standards, selecting the hit compounds for pharmacokinetics and drug-likeness properties and also evaluating the free energies and binding interactions.
C . volubile , phytochemicals were extracted using GC-MS, and their 2D structures were obtained from the NCBI PubChem database. The 3D structures were created with Schrödinger Maestro 11.5’s LigPrep and an OPLS3 force field. Ligand ionization states were created at pH ranges ranging from 7.02.0 (general pH of biological systems). The computation of 25 stereoisomers was left at the specified chiralities (various other chiral centers), and the best potential stereochemical structures were generated per ligand.
The three-dimensional crystal structure of 3E6I was obtained from the RCSB protein data bank. The protein was equipped for calculations using the protein preparation wizard of Schrodinger Maestro 11.5 by filling missing loops and side chains using Prime, assigning bond orders, adding hydrogens, and deleting water beyond 5.00 Å. Using Epik, Tautomeric states of heteroatom groups were generated at a pH of 7.0 ± 2.0, then optimized using PROPKA at pH 7.0. Restrained minimization was carried out by setting heavy atom root-mean-square-deviation to 0.30 Å using an OPLS3 force field.
The maestro receptor grid generation tool was used to generate the receptor grid of CYP2E1, based on the space occupied by its cognate ligands. It is worth noting that the binding pocket of this target protein is located at the X, Y, and Z coordinates.
The compounds generated were subsequently docked in the Maestro’s glide tool, using standard precision SP and extra precision XP algorithm. To obtain probable binding orientations and binding energy of ligands at the specified binding domain of CYP2E1, the grid box with dimensions was used to cover a region on the distal portion of the protein preceded by running of the docking procedure. Following the docking run, compounds that have docking scores (binding energies) lower than the control drug, were exposed to molecular visualization to examine their binding poses (3D) and molecular interaction fingerprints (2D) using PyMOL, subjected to MMGBSA.
ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) is critical at the final stage of the drug discovery and design route to analyze the pharmacokinetics and pharmacodynamics of proposed compounds that could serve as potential drugs [16]. The SWISSADME was used to predict the ADMET characteristics of compounds with the greatest hits following the molecular docking study.
This technique forecasts the free energy of small drug molecules binding to biological macromolecules. The free energy of binding (∆Gbind) was estimated using Prime Schrodinger Maestro 11.5, the OPLS3 force field, and VSGB solvation model with other parameters set to default [17].
The present study employs computational methods such as molecular docking, 2D-Interaction, Prime MMGBSA, and ADMET to discover potential compounds that may exhibit inhibitory effects on the CYP2E1 protein.
Docking simulations were conducted against the target of interest, employing the 49 compounds identified from C . volubile in comparison to Ciprofloxacin, to assess their potential binding affinity to CYP2E1 protein- a therapeutically significant target in hepatotoxicity. The docking outcomes indicated that the compounds exhibit inhibitory potential against the examined protein better than Resveratrol, Sulforaphane, and chlormethiazole standards. The docking analysis depicts the interactions that significantly affect the inhibition (Table 1).
Table 1: Docking scores (kcal/mol) of Hit ligands from Clerodendrum volubile docked against CYP2E1 protein of Cytochrome P450. View Table 1
Analyzing the binding energies reveals that cis , cis -linoleic acid, 3-hydroxy decanoic acid, n-pentadecanoic acid, palmitic acid, and 3,5-dihydroxy-6-methyl-2,3-dihydro-4H-pyran-4-one stand out as the compounds with the most robust inhibitory potentials. They exhibit notable binding affinities against CYP2E1 with energies of -10.746, -10.203, -9.974, -9.177, and -8.447 kcal/mol, respectively. In comparison, several compounds have lower binding energies than the standard Resveratrol with energies of -6.472 kcal/mol respectively. These show that the compounds of C . volubile possess a potential inhibitory effect on the target protein (CYP 2E1).
To further validate the results obtained from the docking procedure, the complexes were subjected to the MM-GBSA post-docking analysis. This analysis is often employed to rigorously assess the stability of complexes formed after docking simulation, by computing the binding free energy of the ligand with the receptor [17]. Intriguingly, results obtained from MM-GBSA analysis have been reported to be correlated with the biological activities derived from experimental procedures [18]. Table 2 presents the results of the MM-GBSA analysis conducted in this study. Interestingly, the compound cis , cis -linoleic acid, which was ranked among the five hit compounds based on the docking results, was found to possess the highest binding free energy measured in kcal/mol -63.43. The other hit compounds which include palmitic acid, n-pentadecanoic acid, 3,5-dihydroxy-6-methyl-2,3-dihydro-4H-pyran-4-one, and 3-hydroxy decanoic acid, have binding free energies of -56.88, -44.63, -38.52, and -24.26 kcal/mol respectively. The standards Resveratrol, Sulforaphane, and chlormethiazole have binding free energies of -41.24, -26.74, and, -12.37 kcal/mol respectively which is lower than the hit compounds this indicates that the hit compounds have better binding strength compared to the standards (the lower the binding energy the better its affinity to the target).
Table 2: Prime MMGBSA of hit Compounds from Clerodendrum volubile against the target protein. View Table 2
These calculations suggest that the enduring and robust complex stability consistently observed with cis , cis -linoleic acid, and palmitic acid across all lead targets implies that the hit compounds might competitively induce prolonged and heightened biological responses compared to standard drugs and other hits.
The diversity in binding scores and affinities among the identified hit compounds implies that these molecules have different degrees of effectiveness in interacting with the target protein. In drug discovery and molecular biology, understanding the interaction between small molecules (such as potential drugs) and specific amino acid residues in target proteins is crucial. The binding affinity refers to the strength of the interaction between a molecule and its target protein, and a higher affinity generally indicates a stronger binding. The figures 1 below show the 2D and 3D interactions of each hit compound and the standards [19] (Table 3).
Table 3: Shows interactions of all ligands’ atoms with amino acid residues in the binding site of CYP2E1 after molecular docking. View Table 3
Figure 1 (1A, 2A, 3A, 4A, 5A, and 6A), indicate the 2D interaction of cis,cis-linoleic acid, 3-hydroxydecanoic acid, n-pentadecanoic acid, palmitic acid, and 3,5-dihydroxy-6-methyl-2,3-dihydro-4H-pyran-4-one and standard respectively to the active site of CYP2e1 receptor while Figure 1 (1B, 2B, 3B, 4B, 5B, and 6B) display their 3D interactions. Notably, the hydrogen bond is represented by a purple arrow, the green region represents the hydrophobic interaction. The interacting residues of the hit compounds and the two standards with the CYP2e1 target protein are displayed in Figure 1 (1A-6A) above.
 Figure 1: The 2D and 3D interaction of the 5 hit compounds using Maestro.
View Figure 1
Figure 1: The 2D and 3D interaction of the 5 hit compounds using Maestro.
View Figure 1
The pharmacokinetic properties of the hit ligands were assessed after evaluating their potency and varied affinities. This investigation is crucial to address issues such as potential side effects, toxicity, limited absorption, and inadequate clearance, commonly encountered during drug development. Understanding these factors are essential to avoid hurdles that often lead to eliminating promising compounds with therapeutic potential. The focus on pharmacokinetics serves as a vital transition from preclinical investigation to clinical trials in the drug development process. The properties of the candidate inhibitors were assessed using SWISSAdme [20], and all components were evaluated for evaluation of critical parameters in the context of drug development. These parameters include the count of hydrogen bond acceptors and donors, molecular weight, and the octanol/water partition coefficient (QPlogPo/w). The evaluation process was thorough, and the results were promising. The key observation is that all the hit ligands, which are potential drug candidates, consistently fall within accepted ranges for these physicochemical attributes. This conformity reaffirms the suitability of these ligands for further consideration and development in the field of drug development. As shown in Table 4, all 5 hit ligands effortlessly met the criteria molecular weight (MW) of ≤ 500 and violated none in Lipinski rule of 5 (Ro5) -one of the rules that determines drug efficacy cis , cis -linoleic acid and palmitic acid violated just one of the 5 rules which is still acceptable. Cytochrome P450 (CYP) is a family of enzymes that catalyze the phase 1 metabolism of xenobiotics at large. Any compound that inhibits selected isoforms would induce a drug-drug interaction. The entire compounds show different inhibitions to the other isoforms of Cytochrome P450 (CYP). They are all moderately soluble and have high GI absorption. The BOILED-egg allows for intuitive evaluation of passive gastrointestinal absorption (HIA) and brain penetration (BBB) in the function of the position of the molecules in the WLOGP-versus-TPSA referential. P-glycoprotein (P-gp), the most extensively studied ATP-binding cassette transporter, functions as a biological barrier by extruding toxic substances and xenobiotics out of cells serving to protect the integrity of tissues and cellular environments (Figure 2 and Figure 3).
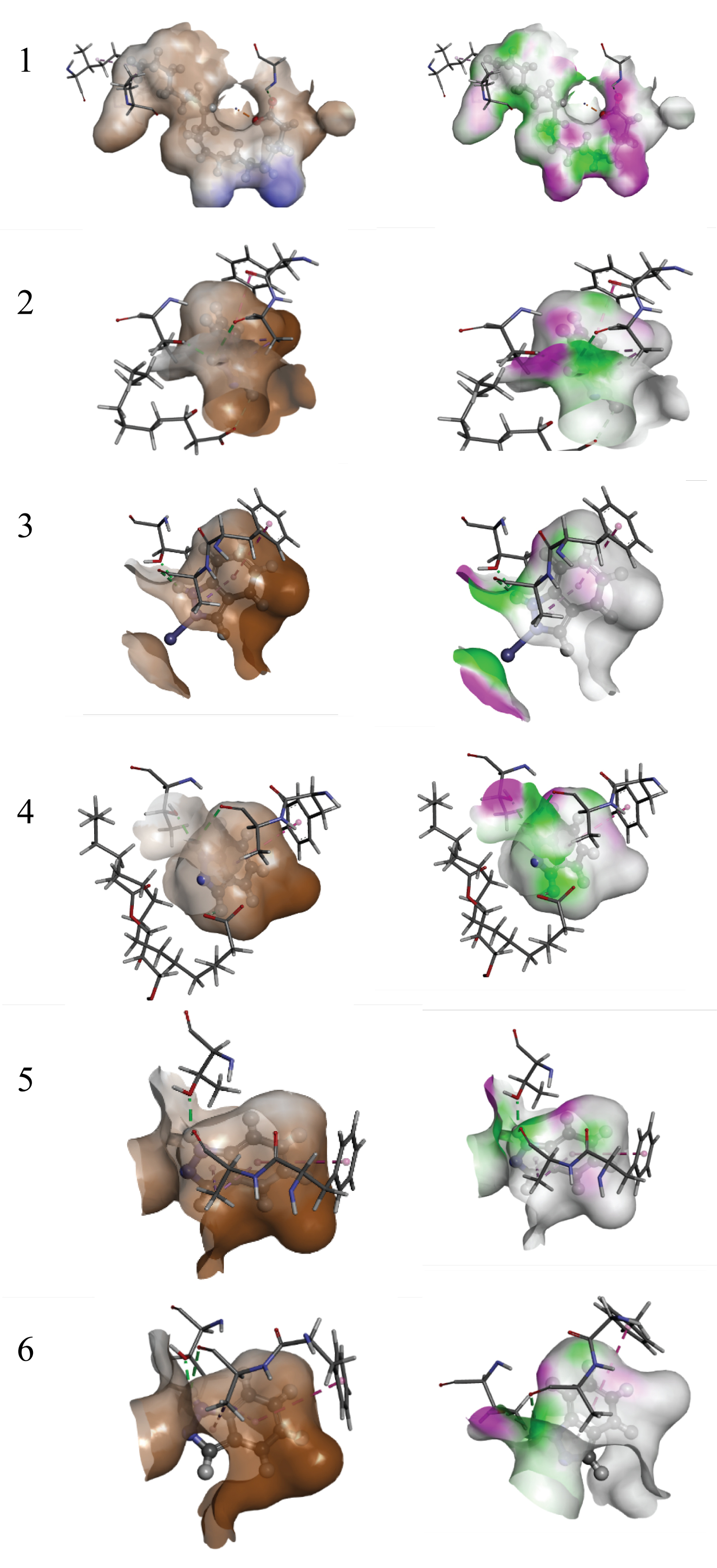 Figure 2: Hydrophobic bonds (X) and Hydrogen bonds (Y) maps between CYP2E1 and five chosen inhibitors including Resveratrol the standard. 1) cis,cis-linoleic acid. 2) 3-hydroxydecanoic acid 3) n-pentadecanoic acid 4) palmitic acid. 5) 3,5-dihydroxy-6-methyl-2,3-dihydro-4H-pyran-4-one 6) Resveratrol. Ligands are presented in ball and stick while amino acid residues of CYP2E1 are in lines executed in BIOVIA’s Discovery Studio 2016.
View Figure 2
Figure 2: Hydrophobic bonds (X) and Hydrogen bonds (Y) maps between CYP2E1 and five chosen inhibitors including Resveratrol the standard. 1) cis,cis-linoleic acid. 2) 3-hydroxydecanoic acid 3) n-pentadecanoic acid 4) palmitic acid. 5) 3,5-dihydroxy-6-methyl-2,3-dihydro-4H-pyran-4-one 6) Resveratrol. Ligands are presented in ball and stick while amino acid residues of CYP2E1 are in lines executed in BIOVIA’s Discovery Studio 2016.
View Figure 2
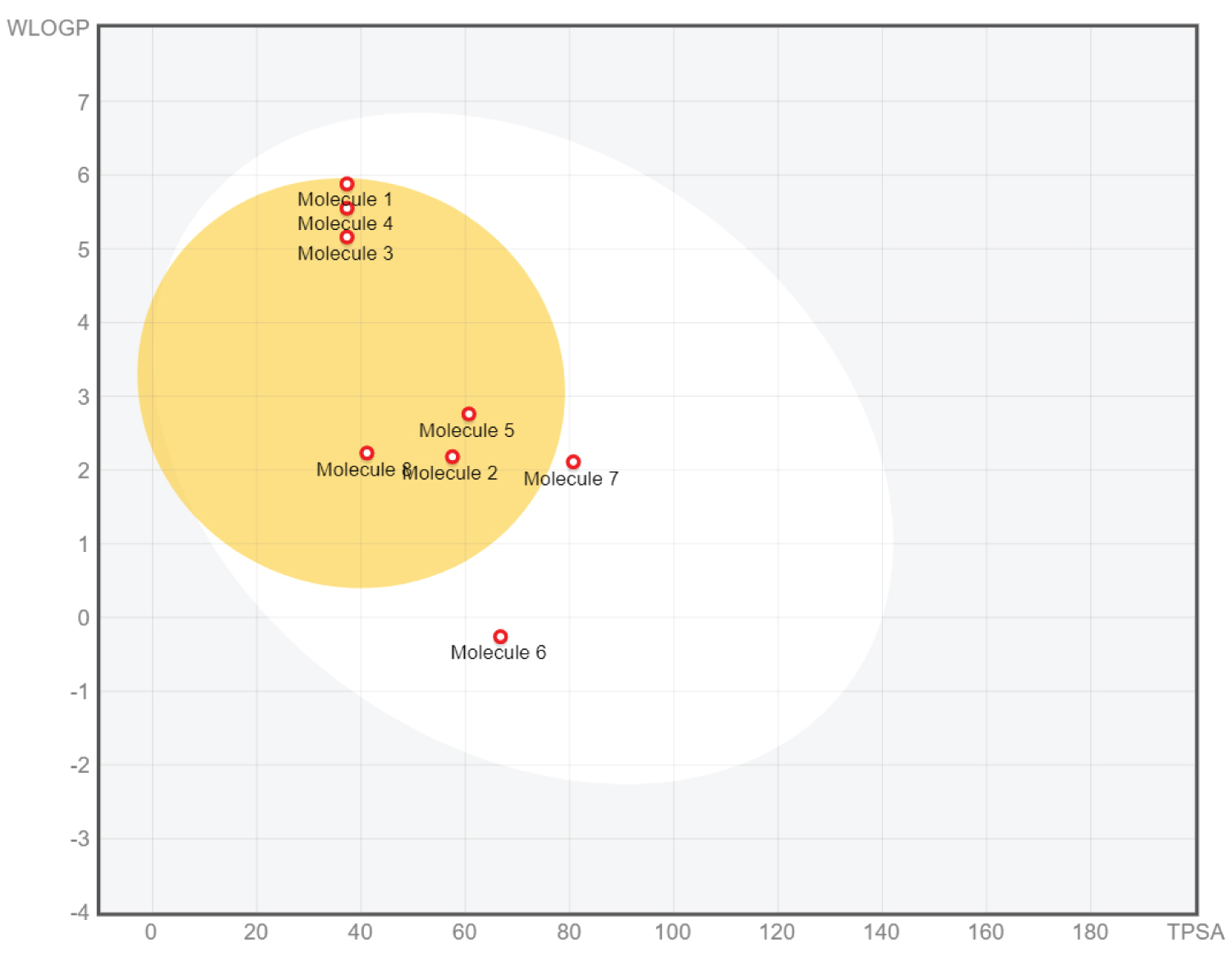 Figure 3: Showing the boiled-egg diagram of the 5 hit compounds and the three standard drugs. The white region is for a high probability of passive absorption by the gastrointestinal tract, and the yellow region (yolk) is for a high probability of brain penetration. Yolk and white areas are not mutually exclusive. In addition, the points are coloured in blue if predicted as actively effluxed by P-gp (PGP+) and in red if predicted as non-substrate of P-gp (PGP−) [21].
View Figure 3
Figure 3: Showing the boiled-egg diagram of the 5 hit compounds and the three standard drugs. The white region is for a high probability of passive absorption by the gastrointestinal tract, and the yellow region (yolk) is for a high probability of brain penetration. Yolk and white areas are not mutually exclusive. In addition, the points are coloured in blue if predicted as actively effluxed by P-gp (PGP+) and in red if predicted as non-substrate of P-gp (PGP−) [21].
View Figure 3
Table 4: ADMET profile and drug likeness of the five hit compounds including the natural substrate using SWISS-ADME server. View Table 4
The study investigated selected natural compounds that have inhibitory and pharmacological properties against the CYP2E1 to reduce the risk of liver diseases, druggable compounds demonstrated a superior binding affinity and favorable ADME profiles using Glide and SWISSADME. The study suggests that molecular docking and simulations are reliable methods for discovering new drugs to combat hepatotoxicity. This highlights the need for novel inhibitors due to adverse effect of existing drugs. These promising compounds should undergo further testing, including atomistic simulation, in - vitro , and in - vivo studies, to validate their potential as effective hepatoprotective drugs.
We have no conflict of interest.
The authors confirm that the data supporting the findings of this study are available within the article.
We received no funding for the research.