Apoptosis is defined as controlled programmed cell death that occurs as physiological process in the development and morphogenesis of multi-cellular organisms, but is also implicated in various diseases, including acute pancreatitis. Apoptosis can be induced by a wide range of stimuli. Using pharmacological inhibitors, our previous results suggest that the cytosolic calcium-dependent cysteine protease calpain-2 plays a crucial role in hydrogen peroxide (H2O2)-induced apoptosis in pancreatic AR42J cells by activating caspases-12, -8 and -3. The present study aimed to corroborate these results using the small interfering RNA (siRNA) technology to knockdown the calpain-2 gene. AR42J cells were incubated with or without calpain-2 siRNA for 48 h, followed by apoptosis induction using H2O2 for 1 h. Calpain-2 downregulation was assessed by immunoblot and immunofluorescence analyses. The effect of calpain-2 downregulation was evaluated by the degree of caspase activation and cell damage. siRNA-induced calpain-2 gene silencing diminished caspase activation and cell damage, leading to an increased viability of the AR42J cells. Our results indicate that calpain-2 plays an important regulatory role in H2O2-induced apoptosis by activating caspases, thus corroborating our previous findings. The molecular mechanisms proposed for H2O2-induced apoptosis in the pancreatic acinar cell model could well also be of great importance in the pathogenesis of acute pancreatitis.
Caspase activation, siRNA, Oxidative stress, Calpain-2, Calpain inhibitor, Calcium, AR42J Cell line
Apoptosis is defined as controlled programmed cell death occurring as physiological process in the development and morphogenesis of multi-cellular organisms. It is induced to eliminate genetically altered or injured cells without ensuing inflammation. There are two different forms of apoptosis: the extrinsic (death receptor-mediated) and the intrinsic (mitochondrial) pathway. One of the earliest events in apoptosis is the activation of cytosolic cysteine proteases, the caspases. Caspases are classified into two three major groups, the upstream initiator caspases (caspases-2, -8, -9, -10), the downstream executioner caspases (caspase-3, -6 and -7) and the inflammatory caspases (-1, -4, -5 and -11). Apoptosis can be induced by a wide variety of stimuli such as heat, radiation, hypoxia and cytotoxic anticancer drugs. In addition, also moderate oxidative stress leads to apoptosis [1,2]. Oxidative stress occurs when the concentration of reactive oxygen species that are regularly formed during oxidative cellular metabolism exceeds the capacity of the antioxidant defense system that naturally neutralizes and eliminates them. Subsequently, this can lead to cell damage or cell death [3]. In pancreatic acinar cells, oxidative stress causes a sustained increase in the cytosolic calcium (Ca2+) concentration that precedes morphological and functional damage [4-6]. One early event following the intracellular Ca2+ accumulation is an activation of calpain [7]. Calpains are Ca2+-dependent cytosolic cysteine proteases that belong to the papain family. Beside several tissue-specific forms (n-calpains), two ubiquitous isoforms, calpain-1 (μ-calpain) and calpain-2 (m-calpain), have been described that differ in their Ca2+ requirement for in vitro activation. Unlike digestive proteases such as trypsin or carboxypeptidases, which catalyze degradation of their substrates, calpains cause limited proteolysis of their substrates, leading to distinct peptide fragments. Regulated proteolysis by calpains is required for various physiological processes, including cytoskeletal remodeling, membrane fusion, cell migration, proliferation and apoptosis [8]. Recently, we have shown that calpain-2 is activated by hydrogen peroxide (H2O2)-induced oxidative stress, leading to apoptosis in rat pancreatic AR42J cells. Investigating the mechanism by which calpain-2 may contribute to acinar cell apoptosis, our results showed an activation of caspase-12, caspase-8 and caspase-3 [9]. Since the pharmacological inhibitors used to establish the above hypothesis are not absolutely specific for calpain [10], in the present study, we aimed to confirm our previous results. Exploiting small interfering RNA (siRNA) technology to knockdown the calpain-2 gene, which allows linking the findings of the study to calpain-2 protein enabled us to pin-point the enzyme's key regulatory role in the mechanism of apoptosis. siRNA consists of a RNA duplex with length between 19 and 21 bases, and degrades target messenger RNAs via the RNAi pathway, thus leading to downregulation of target gene expression [11-13].
Our findings confirm that apoptosis in pancreatic AR42J cells observed in response to H2O2-induced oxidative stress may require activation of calpain-2, thus corroborating our previous conclusions [9].
Accutase was purchased from Biolegend (San Diego, CA, USA). Heat inactivated Fetal Bovine Serum (FBS) and Dulbecco's Modified Eagle's Medium (DMEM) were obtained from Life Technologies (Eggenstein, Germany). Penicillin and streptomycin were procured from Biochrom Seromed (Berlin, Germany). Dexamethasone (Fortecotin Mono-4) was obtained from Merck (Darmstadt, Germany). Dako Antibody Diluent, DakoCytomation Fluorescent Mounting Medium and Dako Protein Block were purchased from Dako Deutschland (Hamburg, Germany). Calpain-2 siRNA (sc-60100), control siRNA-A (scrRNA) (sc-37007) and RNA Diluent RNase-free H2O were procured from Santa Cruz Biotechnology (Dallas, TX, USA). The siRNA product consists of three target-specific 19-25 nucleotide-long double stranded RNA molecules with 2-nt 3' overhangs on each end of a strand. The sequence of the siRNA has not been disclosed by the company. HiPerFect® Transfection Reagent (HPF) was purchased from Qiagen (Hilden, Germany). Goat serum and H2O2 were obtained from Sigma-Aldrich (Deisenhofen, Germany). Tris-Glycine-SDS buffer and Towbin buffer were purchased from Serva (Heidelberg, Germany). Tris-Glycin gradient gels (8%-16%) were obtained from biostep (Burkhardtsdorf, Germany). SR-FLICA apoptosis detection kit was bought from Immunochemistry Technologies (Bloomington, MN, USA).
Rabbit polyclonal calpain-2 antibodies from Santa Cruz Biotechnology (H-240; Dallas, TX, USA) and Millipore Corporation, (AB 81023; Temecula, CA, USA) were used. Alexa Fluor® 488 goat anti-rabbit IgG (H + L) was obtained from Life Technologies (Eggenstein, Germany). Goat-anti rabbit IgG (H + L) secondary antibody, HRP conjugate was purchased from Thermo Fisher Scientific (Dreieich, Germany).
The rat pancreatic AR42J cell line (American Type Culture Collection, Rockville, MD, USA) was used for the present experiments. Dexamethasone treatment has been found to convert these cells into exocrine cells [14]. AR42J is as yet the only cell line that exhibits many characteristics of normal pancreatic acinar cells such as synthesis and secretion of digestive enzymes. Thus, this cell line is predestinated for the investigation of secretion, growth, proliferation, and oxidant-induced apoptosis of exocrine pancreatic cells [15].
Cells were seeded at a density of 6 × 104/cm2 in 75 cm2 culture flasks and routinely grown in DMEM supplemented with 10% (v/v) FBS, 50 U/ml penicillin and 50 μg/ml streptomycin at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. When reaching confluence, the cells were detached by accutase according to the manufacturer's recommendation.
AR42J cells were used between passages 7 and 13. For each group, including the controls, cells were suspended in 2 ml DMEM, which contained FBS and antibiotics, and added to 24-well plates (triplicates, density of 8 × 104 cells/well). Cells were incubated under standard cell culture conditions. After reaching 70% subconfluence, the medium was replaced by 500 μl fresh DMEM. For the calpain-2 knockdown, 0.002 nmol calpain-2 siRNA was diluted in 100 μl DMEM and mixed with 3 μl and 1.5 μl HPF, respectively. While the solution incubated at room temperature for 10 min, the transfection complexes were formed and subsequently were added drop-wise onto the cells, resulting in a final siRNA concentration of 3.33 nM. Control cells were incubated with control siRNA (scrRNA) at the same concentration and dilution, but without gene silencing effect on calpain-2 or with DMEM only. Thereafter, the cells were incubated under standard cell culture conditions. In order to induce cell differentiation, 100 nM dexamethasone were added to all samples 6 h after transfection. After further 48 h, a part of the siRNA-treated cells and untreated control cells were harvested for Western blot analysis to investigate the calpain-2 knockdown at the protein level. Additionally, cells were used for apoptosis experiments. siRNA-treated cells and control cells were exposed to H2O2 (final concentration: 250 μM). The medium was removed and replaced by DMEM 1 h after H2O2 addition. Cells without H2O2 exposure served as control. Then, the cells were incubated for further 5 h.
Cell damage was assessed by measuring the release of lactate dehydrogenase (LDH) into the culture medium using the UniCel® DxC 800 Synchron Clinical System from Beckman Coulter (Krefeld, Germany). In addition, the trypan blue exclusion assay was used to calculate the cell viability. Briefly, at the end of the experiments, cells were harvested using accutase and an aliquot of the cell suspension was diluted 1:1 with 0.4% trypan blue solution. Cell number was determined using a Neubauer chamber. All cells excluding trypan blue were considered viable.
The cells were lysed in 200 μl buffer containing 50 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 5 mM EGTA, 25 mM NaF, 25 mM β-Glycerophosphat, 0.3% NP40, and 0.2% Triton X-100 supplemented with the protease inhibitors Pefabloc (2 mM) and Leupeptin (0.011 mM), and kept on ice for 10 min. Thereafter, the samples were stored at -80 °C until use.
For immunoblotting, samples were defrosted on ice, sonified for 15 s and centrifuged at 4 °C for 20 min. The protein concentration in the supernatant was determined using the Pierces BCA Protein Assay Kit from Life Technologies (Rockford, Il, USA). Thereafter, protein samples were mixed with sample buffer Roti-Load 1 (Carl Roth, Karlsruhe, Germany) according to the manufacturer's suggested protocol, heated at 95 °C for 4 min and subjected to Laemmli SDS-PAGE. The fractionated proteins were electrophoretically transferred onto PVDF membranes overnight at 25 V using Towbin's buffer. Then the membranes were incubated with the blocking buffer TBS-T containing 5% goat serum at room temperature for 60 min and probed with the respective primary antibody overnight at 4 °C. Labeled proteins were visualized by enhanced chemiluminescence using horseradish peroxidase-conjugated secondary antibody supplied with the Pierce™ Fast Western Blot Kit (Thermo Fisher Scientific) at a 1:20,000 dilution. The densitometric quantification was carried out using the software ImageJ.
Cytospin cell preparations were centrifuged on Superfrost slides (Merck, Germany). Slides were air-dried for 24 h and stored at -80 °C. The sections were fixed with 4% methanol at -20 °C for 10 min and permeabilized with 0.1% Triton X-100 dissolved in PBS for 10 min at room temperature. Nonspecific antibody binding was blocked with Dako Protein Block for 20 min. Thereafter, the cells were incubated with the rabbit polyclonal calpain-2 antibody (H-240; 1:50 dilution) in a humid and dark chamber for 60 min. After four washing cycles with PBS-T containing 1% FBS, the cells were treated with anti-rabbit IgG (H + L) F(ab')2 fragment conjugated to Alexa Fluor® 488 fluorescent dye (1:200) for 30 min at room temperature. To ensure specificity of the immune reaction, cells were incubated with the secondary antibody only under otherwise identical conditions. The slides were examined using the fluorescence microscope EUROStar III Plus (Euroimmun, Luebeck, Germany; excitation/emission: 493/516 nm, equipped with the software Europicture). The evaluation of the fluorescence intensity was performed using a scoring system and the ImageJ software. The Corrected Total Cell Fluorescence (CTCF) of 50 cells per slide was calculated [16]. A fluorescence intensity until 10,000 was valuated with 1, values ranging from 10,000 to 20,000 were assessed with 2, values ranging from 20,000 to 30,000 were assessed with 3 and values above 30,000 were assessed with 4.
Apoptosis was detected using the SR-FLICA (sulforhodamine Fluorescent Labeled Inhibitors of Caspases) apoptosis detection kit containing the cell permeable and non-toxic SR-FLICA inhibitor, SR-VAD-FMK (sulforhodaminyl-L-valylalanylaspartyl fluoromethyl ketone, and the cell permeable DNA binding dye, Hoechst 33342. The FLICA reagent reacts covalently with activated caspases and is retained in apoptotic cells, while unbound reagent will diffuse out of the cell. After transfection with calpain-2 siRNA and induction of oxidative stress as described above, the cells were incubated with FLICA solution at 37 °C in 5% CO2 environment for 30 min. The medium was removed and 1.5 μl Hoechst stain (0.5% v/v) were added followed by additional 5 min incubation. Control cells were incubated with or without H2O2, whereas a further aliquot of cells was treated with both scrambled siRNA and H2O2. Finally, the cells were evaluated using a fluorescence microscope Nikon Eclipse E600 (excitation 550 nm, emission 590 nm for red fluorescence, and excitation 365 nm, emission 480 nm for Hoechst stain). The intensity of the red fluorescence correlates with the concentration of activated caspases.
Results are expressed as mean + SEM. To compare data, One Way Analysis of Variance (ANOVA) was performed by using the statistical software package SigmaStat 3.5 from Jandel Corporation (Erkrath, Germany). P values of < 0.05 were considered as statistically significant.
To investigate the role of calpain-2 in H2O2-induced AR42J cell damage, expression of the calpain-2 gene was downregulated using calpain-2 siRNA. A successful protein knockdown depends on the concentration of siRNA and the ratio of the transfection reagent and siRNA concentration. In the first step, we transfected the cells with 100 μl of 10 nM siRNA mixed with 3 μl transfection reagent HiPerFect® (HPF) in dependence on the incubation time (24 h, 30 h, and 48 h). Western Blot analyses of calpain-2 and evaluation of fluorescence micrographs using a scoring system showed only an insufficient inhibitory effect after 24 h und 30 h (~ 7%) (data not shown). Next, the experiments were repeated with higher siRNA concentrations (20 nM, 50 nM, and 100 nM). The concentrations of 20 nM and 50 nM resulted in a calpain-2 downregulation of approximately 25%, whereas in response to 100 nM siRNA, the transfection effects were smaller than 25% (data not shown). In the next step, the cell to lipid-carrier ratio was modified by reducing the volume of HPF. Thus, siRNA (10 nM, 20 nM, 50 nM and 100 nM) was dissolved in 1.5 μl HPF followed by incubation with the cells for 24 h, 48 h and 72 h. The highest calpain-2 downregulation was observed in response to 20 nM siRNA after 24 h and 48 h (Figure 1). In further experiments, we compared the effect of 20 nM siRNA dissolved in 1.5 μl or 3 μl HPF in dependence on the incubation time of 24 h and 48 h, respectively. As shown by western blot analyses and densitometric evaluation of the blots, the calpain-2 expression decreased by 20 ± 7% and 60 ± 11% after 24 h (P < 0.01) and 48 h (P < 0.05), respectively, compared with the control (Figure 2A and Figure 2C, compare lanes 1 and 3 each; Figure 2B and Figure 2D). Based on these results, 20 nM calpain-2 siRNA dissolved in 1.5 μl HPF for 48 h were applied to evaluate the role of calpain-2 in oxidant-induced AR42J cell apoptosis. In several experiments, the effect of this approach was confirmed as shown exemplary in Figure 3A (compare lane 1 with lanes 3 and 5). The results also showed that both controls, DMEM and scrRNA, exerted no effect on the calpain-2 expression (Figure 3A, lanes 3 and 5).
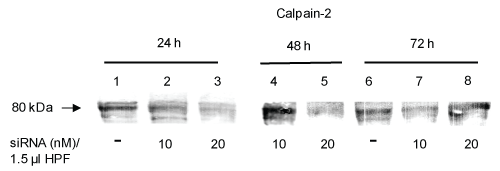 Figure 1: siRNA-induced calpain-2 gene silencing in rat pancreatic AR42J cells in dependence on the siRNA concentration and the incubation time. Cells were incubated with 10 nM or 20 nM calpain-2 siRNA dissolved in 1.5 μl transfection reagent HPF for 24 h, 48 h or 72 h, respectively. Control cells were incubated with cell culture medium. The protein expression of calpain-2 was evaluated by immunoblotting. Representative Western blot of calpain-2 (10 μg) probed with the polyclonal anti-calpain-2 antibody AB 81023 (1:1000) of two independent experiments. View Figure 1
Figure 1: siRNA-induced calpain-2 gene silencing in rat pancreatic AR42J cells in dependence on the siRNA concentration and the incubation time. Cells were incubated with 10 nM or 20 nM calpain-2 siRNA dissolved in 1.5 μl transfection reagent HPF for 24 h, 48 h or 72 h, respectively. Control cells were incubated with cell culture medium. The protein expression of calpain-2 was evaluated by immunoblotting. Representative Western blot of calpain-2 (10 μg) probed with the polyclonal anti-calpain-2 antibody AB 81023 (1:1000) of two independent experiments. View Figure 1
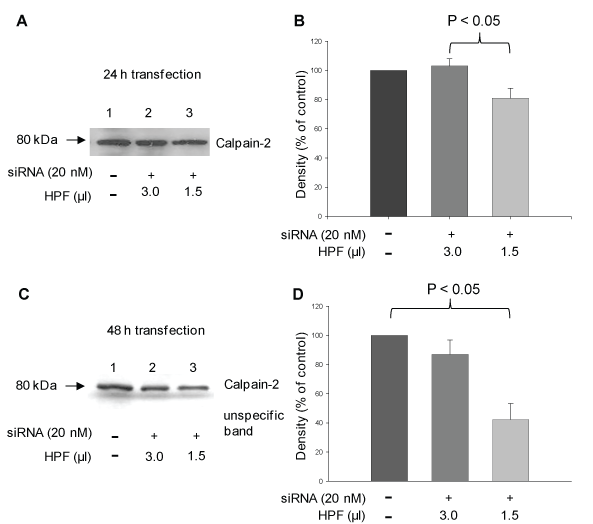 Figure 2: siRNA-induced calpain-2 gene silencing in rat pancreatic AR42J cells in dependence on the volume of the transfection reagent HPF and incubation time. Cells were incubated with 20 nM calpain-2 siRNA dissolved in 1.5 μl or 3 μl transfection reagent HPF for 24 h or 48 h. Control cells were incubated with cell culture medium (A) Representative Western blot of calpain-2 after 24 h (20 μg protein) and (C) after 48 h (15 μg protein) probed with polyclonal anti-calpain-2 antibody H-240 (1:200) and polyclonal anti-calpain-2 antibody AB 81023 (1:1000), respectively, including the corresponding densitometric quantification of five (B) and four (D), respectively, independent experiments. The unspecific band in Figure 2C was used as loading control. The results of the densitometric measurements are expressed as mean + SEM. Statistical comparisons were performed using ANOVA followed by Tukey test. P values of < 0.05 were considered as statistically significant. View Figure 2
Figure 2: siRNA-induced calpain-2 gene silencing in rat pancreatic AR42J cells in dependence on the volume of the transfection reagent HPF and incubation time. Cells were incubated with 20 nM calpain-2 siRNA dissolved in 1.5 μl or 3 μl transfection reagent HPF for 24 h or 48 h. Control cells were incubated with cell culture medium (A) Representative Western blot of calpain-2 after 24 h (20 μg protein) and (C) after 48 h (15 μg protein) probed with polyclonal anti-calpain-2 antibody H-240 (1:200) and polyclonal anti-calpain-2 antibody AB 81023 (1:1000), respectively, including the corresponding densitometric quantification of five (B) and four (D), respectively, independent experiments. The unspecific band in Figure 2C was used as loading control. The results of the densitometric measurements are expressed as mean + SEM. Statistical comparisons were performed using ANOVA followed by Tukey test. P values of < 0.05 were considered as statistically significant. View Figure 2
Incubation of transfected cells with H2O2 led to a clearly decrease in the calpain-2 expression compared with the controls, DMEM and scrRNA, and cells incubated with H2O2 alone (Figure 3A, compare lane 2 with lanes 3, 4 and 5). These results were supported by the immunofluorescence microscopic evaluation. As demonstrated in Figure 3B, the siRNA-transfected AR42J cells were characterized by a weak calpain immunostaining, whereas the control cells were brightly green stained. Most of the calpain was found within the cytosol, whereas a smaller part was localized beneath the cell membrane. In response to H2O2 alone, the calpain immunostaining was similar to that of the control cells. These results were supported by evaluation of the fluorescence intensity using a scoring system as described in the Methods section. In fact, the fluorescence intensity of the transfected cells was decreased by 30 ± 6% compared with the control.
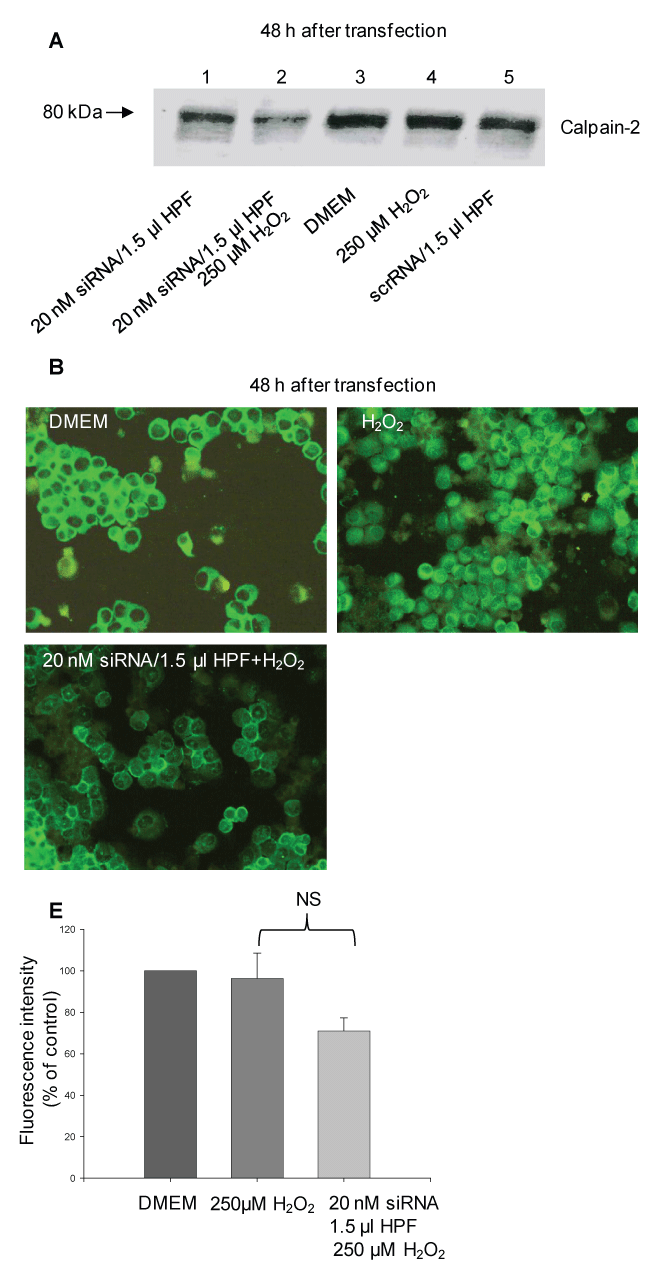 Figure 3: siRNA-induced gene knockdown of calpain-2 in H2O2-treated rat pancreatic AR42J cells. At 48 h after transfection using 20 nM siRNA/1.5 μl HPF, the cells were incubated in serum-free DMEM and exposed to 250 μM H2O2 (20 nM siRNA/1.5 μl HPF and H2O2). After 1 h, the cells were incubated in serum-containing DMEM for further 5 h. Control cells were treated with the transfection complex alone (20 nM siRNA/1.5 μl HPF), incubated in cell culture medium (control), H2O2 and scrRNA (srcRNA/1.5 μl HPF), respectively. (A) Representative Western blot of calpain-2 (20 μg) of three independent experiments at 48 h after transfection probed with the polyclonal anti-calpain-2 antibody (AB 81023; 1:1000); (B) Representative fluorescence micrographs probed with rabbit polyclonal anti-calpain-2 antibody (H-240; 1:50) and stained with anti-rabbit IgG (H + L) F(ab')2 fragment conjugated to Alexa Fluor® 488. Original magnification ×20; n = 3-4; (E) Fluorescence intensity of the fluorescence micrographs was performed using a scoring system and the ImageJ software. Statistical comparisons were performed using ANOVA. NS: non-significant. View Figure 3
Figure 3: siRNA-induced gene knockdown of calpain-2 in H2O2-treated rat pancreatic AR42J cells. At 48 h after transfection using 20 nM siRNA/1.5 μl HPF, the cells were incubated in serum-free DMEM and exposed to 250 μM H2O2 (20 nM siRNA/1.5 μl HPF and H2O2). After 1 h, the cells were incubated in serum-containing DMEM for further 5 h. Control cells were treated with the transfection complex alone (20 nM siRNA/1.5 μl HPF), incubated in cell culture medium (control), H2O2 and scrRNA (srcRNA/1.5 μl HPF), respectively. (A) Representative Western blot of calpain-2 (20 μg) of three independent experiments at 48 h after transfection probed with the polyclonal anti-calpain-2 antibody (AB 81023; 1:1000); (B) Representative fluorescence micrographs probed with rabbit polyclonal anti-calpain-2 antibody (H-240; 1:50) and stained with anti-rabbit IgG (H + L) F(ab')2 fragment conjugated to Alexa Fluor® 488. Original magnification ×20; n = 3-4; (E) Fluorescence intensity of the fluorescence micrographs was performed using a scoring system and the ImageJ software. Statistical comparisons were performed using ANOVA. NS: non-significant. View Figure 3
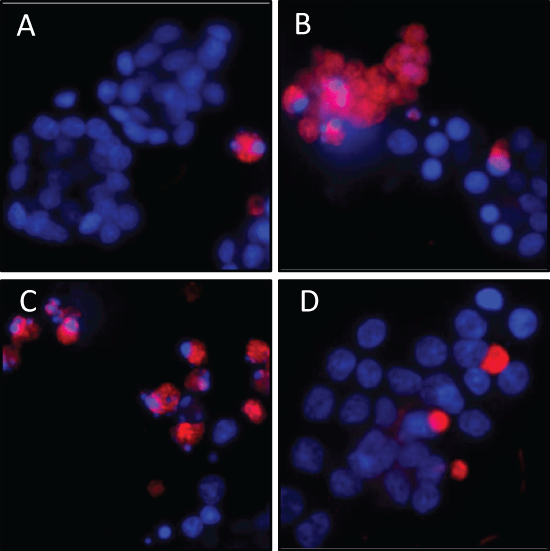 Figure 4: Reduction of H2O2-induced apoptosis in rat pancreatic AR42J cells by siRNA-induced calpain-2 gene silencing. Experimental design as in Figure 3. The cells were stained with sulforhodamine-labeled caspase inhibitor SR-VAD-FMK and Hoechst 33342 to label nuclear DNA. Shown are representative fluorescence micrographs of three independent experiments: (A) scrRNA-treated control cells; (B) H2O2-treated cells; (C) H2O2-treated cells in the presence of scrRNA and (D) H2O2-treated cells in the presence of siRNA. Original magnification ×20. View Figure 4
Figure 4: Reduction of H2O2-induced apoptosis in rat pancreatic AR42J cells by siRNA-induced calpain-2 gene silencing. Experimental design as in Figure 3. The cells were stained with sulforhodamine-labeled caspase inhibitor SR-VAD-FMK and Hoechst 33342 to label nuclear DNA. Shown are representative fluorescence micrographs of three independent experiments: (A) scrRNA-treated control cells; (B) H2O2-treated cells; (C) H2O2-treated cells in the presence of scrRNA and (D) H2O2-treated cells in the presence of siRNA. Original magnification ×20. View Figure 4
To detect the role of calpain-2 in H2O2-induced apoptosis, the activation of caspases was evaluated using the SR-FLICA multi-caspases assay. The intensity of the cellular red fluorescence emitted from the fluorescencing inhibitor bound to activated caspases correlates with the caspase concentration. The fluorescence micrographs showed only few apoptotic cells in the control population (Figure 4A), whereas in H2O2 exposed cells, a larger number of red-fluorescent cells were visible (Figure 3B). Similarly, in the population treated with scrambled siRNA (scrRNA) before H2O2 administration, many apoptotic cells were visible (Figure 4C). On the other hand, transfection with calpain-2 siRNA clearly reduced H2O2-induced caspase activation in the AR42J cells for the most part, indicating reduced apoptosis (Figure 4D).
Treatment of cells with 250 μM H2O2 for 1 h led to cell damage as observed after 5 h. Thus, cellular viability was significantly decreased by 33 ± 6% compared with the control (P < 0.01) (Figure 5A). Transfection of the cells with calpain-2 siRNA before exposure to H2O2 greatly prevented the decrease in viability. Indeed, there was no significant difference in the viability between control cells and transfected H2O2-treated cells.
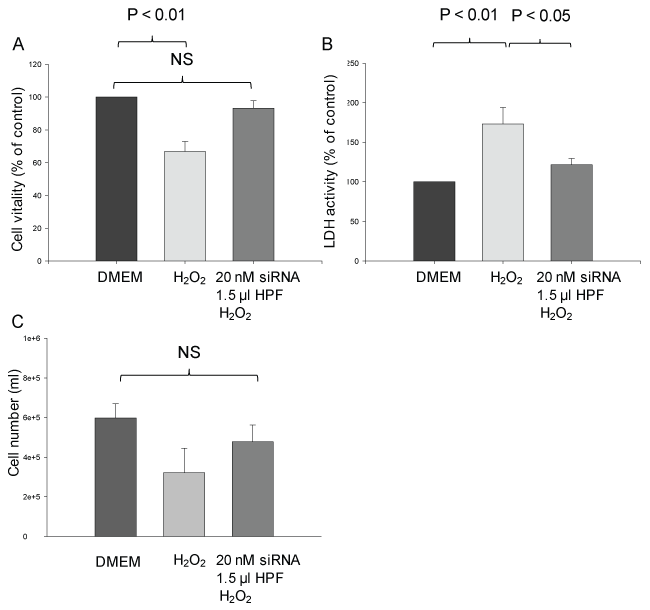 Figure 5: Effect of siRNA-induced calpain-2 gene silencing on H2O2-induced damage of rat pancreatic AR42J cells. Experimental design as in Figure 3. Cell viability (A), LDH release (B) and cell number (C) were elevated at 6 h after H2O2 addition. The results are expressed as mean + SEM (n = 4-5). NS: non-significant. Statistical comparisons were performed using ANOVA followed by Tukey test. P values of < 0.05 were considered as statistically significant. View Figure 5
Figure 5: Effect of siRNA-induced calpain-2 gene silencing on H2O2-induced damage of rat pancreatic AR42J cells. Experimental design as in Figure 3. Cell viability (A), LDH release (B) and cell number (C) were elevated at 6 h after H2O2 addition. The results are expressed as mean + SEM (n = 4-5). NS: non-significant. Statistical comparisons were performed using ANOVA followed by Tukey test. P values of < 0.05 were considered as statistically significant. View Figure 5
The LDH activity released into the incubation medium, a further marker of cell damage, was enhanced by 73 ± 20% in response to H2O2 compared with the control (P < 0.01) (Figure 5B). However, when the cells were transfected with calpain-2 siRNA before H2O2 treatment, the LDH was increased by 22 ± 8% only (P < 0.05).
H2O2-induced cell damage is also characterized by a decrease in the cell number that was reduced by ~ 50% compared with the control. Transfection of the cells with calpain-2 siRNA exerted a protective effect, leading to a decrease in the cell number by 20% when compared with the control (Figure 5C).
siRNA technology has been established as a powerful experimental tool to investigate gene functions in vitro and in vivo. By producing loss-of-function phenotypes, the technology allows linking experimental results to specific genes [17,18]. We used siRNA-mediated gene knockdown to corroborate our previous conclusions, which assign to calpain-2 an important role in the apoptosis of pancreatic AR42J cells induced by oxidative stress [9].
As a prerequisite for a successful siRNA-mediated downregulation of genes the RNAi pathway has been identified, which functions in a variety of cell lines in a species-specific manner [19,20]. Our present results, demonstrating the downregulation of calpain-2 in AR42J cells, indicate that the RNAi mechanism exists in this cell type as well. In support of our findings, further genes have been successfully silenced in AR42J cells, including the gene of phospholipase II, Protein Inhibitor of Activated Signal Transducer (PIASI), RAB8, pancreatitis-associated proteins and of the Translocating Chain-Associated Membrane protein 1 (TRAM1) [21-25].
In general, siRNAs cause a satisfactory decrease in the target mRNA levels within 18 h or less. However, AR42J cells are slowly growing cells and need longer times to reach an optimum expression profile than actively growing ones. Treatment with dexamethasone to induce AR42J cell differentiation further slows down their growth [14]. Furthermore, it seems to be difficult to effectively downregulate proteins with a long half-life by transient siRNA transfection. There is evidence that stable proteins require longer times of exposure to siRNAs in order to be knocked down compared to less stable ones. Both ubiquitous calpain isoforms have metabolic half-lives of approximately five days [26,27], which accords with the optimal calpain-2 downregulation after 48 h in our experiments. In line with these findings, calpain-2 downregulation has been reported to be achieved after 24 or 48 h in various types of cellular systems, including rheumatoid synovial cells, human hepatoma cells, osteosarcoma cells and cardiomyocytes [28-31].
A further important factor that influences efficient protein downregulation is the gene-silencing potency of the siRNA [19]. To knockdown calpain-2, we used calpain-2 siRNA consisting of a pool of three target-specific 19-25 nucleotide-long double stranded RNA molecules with 2-nt 3' overhangs on each end of the strand. Pooling of siRNAs allows reducing non-specific gene modulation [20]. Furthermore, the success of transfection depends on the optimal delivery conditions, which are specific for the cell type being used [21]. In vitro, cationic lipids have been reported to transport nucleic acids across membranes into cells with high efficiency [22]. Therefore, we have chosen a non-toxic lipid-based transfection reagent for the present study. Optimizing the cells to lipid-carrier ratio for efficient transfection, we found that a higher ratio using 1.5 μl transfection reagent resulted in higher protein downregulation than the lower one of 3 μl. Our transfection experiments using 20 nM calpain-2 siRNA and 1.5 μl transfection reagent resulted in a calpain protein downregulation of 60% after 48 h. The low final siRNA concentration of 3.3 nM leading to this result indicates that the siRNA used possesses high gene-silencing activity. Indeed, active siRNAs have been reported to exhibit IC50 values in the range of 0.2-0.5 nM, and 1-10 nM siRNA are sufficient to achieve an effective downregulation of target genes in cell cultures [11,23].
Previously, we have shown that exposure of AR42J cells to moderate H2O2-induced oxidative stress caused activation of calpain-2 via a pathological Ca2+ increase. Enhancement of calpain-2 activity was followed by activation of the caspases-12 and -8 and, subsequently, of caspase-3, which executes apoptosis. Our conclusion that calpain activation plays a crucial role in the H2O2-induced apoptotic pathway was partially based on Western blot analysis of calpain-2 and its degradation products. In addition, we demonstrated that blocking of calpain by the pharmacological inhibitor PD150606 significantly reduced activation of the caspases and cell damage [9]. In the present study, we were able to confirm these results by downregulating the protease using siRNA knockdown technology. In accordance with the proposed pathway of H2O2-induced apoptosis in AR42J cells [9], siRNA-induced downregulation of calpain-2 correlated with an increased viability of cells. Our results thus corroborate the key regulatory role of calpain-2 in this pathway. In addition, we show that beside caspases a second proteolytic system, the calpain system, may play a role in the regulation of H2O2-induced apoptosis. Oxidative stress has been accepted to be an important event in the pathogenesis of acute pancreatitis [32]. We, therefore, assume that the molecular mechanism of apoptotic cell death investigated in the present study could also be of importance in acute pancreatitis.
The authors thank Juliane Dietrich and Saskia Krohn for their excellent technical assistance.
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.