Retinoids and its derivatives are known to regulate tumor progression. Our previous study in Colorectal Cancer (CRC) has shown that the expression of LRAT, a gene converts excess retinol into retinyl ester to balance retinoids homeostasis, may be regulated by its promoter methylation status to modulate the retinoids synthesis. In this report, we begin to explore the potential mechanism of LRAT mediated retinoid metabolism. Our data indicate CRC patient of LRAT hypermethylation associated with better prognosis. A consistent finding is shown in siRNA mediated LRAT silencing, which leads to slow growth of CRC cell lines. We have also observed favorable CRC prognosis occurred in patients of both LRAT and RAR beta hypermethylation, suggesting the better CRC prognosis may be mediated through RAR beta independent pathway.
CpG methylation, Thermostable ligase, Colorectal cancer, LRAT
LRAT: Lecithin Retinol Acyltransferase; RAR: Retinoic Acid Receptor
The retinoids, a group of compounds that include retinol (vitamin A) and its metabolites, play a fundamental role in normal cell growth, differentiation and apoptosis [1-4]. Retinol is regularly obtained from the diet. In animal products such as meat and eggs, retinol is stored in the form of retinyl esters. Upon digestion, the retinyl esters are released and subsequently hydrolyzed to retinol. Retinol can also be synthesized from the pro-vitamin A carotenoids (β-carotene, α-carotene and β-cryptoxanthin) found in fruits and vegetables such as carrots, cantaloupe, broccoli and dark green leafy vegetables. In the cytosol, retinol is oxidized to retinaldehyde by Retinol Hydrogenases (RDHs) or Alcohol Dehydrogenases (ADHs) and then to retinoic acid by Retinaldehyde Dehydrogenases (RALDHs). Excess of retinol can be converted to retinyl esters by the enzyme Lecithin:Retinol Acetyltransferases (LRAT) and stored in the liver.
Colorectal Cancer (CRC) is one of the leading causes of cancer related deaths. Studies have shown altering retinol metabolism may have chemotherapic benefits on CRC [1,5-8]. For example, the most active metabolite of retinol, retinoic acid, is transported to cell nucleus where it binds to the Retinoic Acid Receptors (RAR α,β,γ), stimulates downstream gene expression, affects cell cycle progression, and leads to cancer cell growth inhibition. The challenge of using retinoic acid in cancer chemotherapy is that the expression of RARs is often lost at advanced tumor stages, rendering the poor efficacy of retinoic acid treatment. This phenomenon of retinoic acid resistance has been shown partly due to epigenetic changes such as aberrant histone modification and DNA methylation to silence RAR gene expression.
Previously, we have shown frequent LRAT hypermethylation in earlier (I/II) than in later (III/IV) CRC stages [9]. This inverse relationship between CRC stages and the hypermethylated LRAT instances is a tumor-specific, non-random event (p < 0.0001). We have also demonstrated that LRAT hypermethylation is independent of tumor MSI status; therefore, this LRAT methylation pattern is not a typical MSI feature of possessing many hypermethylated genomic loci. In this study, we begin to explore the aberrant LRAT hypermethylation and its relationship to CRC clinical outcomes.
In many cancers, the majority of aberrant promoter hypermethylation are positively correlated with tumor stages [10]. Namely, the high instances of hypermethylated promoters are more often seen in late tumor stages, such as RAR promoter hypermethylation seen in CRC. Since patients with early-stage CRC are known to have better outcomes [11], and the high instances of LRAT hypermethylation occurs in early-stage CRC, one may predict a favorable prognosis of LRAT hypermethylated CRC cases. As shown in Figure 1A, Kaplan-Meier survival analysis was performed on 123 CRC cases with known LRAT methylation status. All these samples are microsatellite stable and none colonic polyps, ensuring the survival analysis is not biased towards a better outcome (e.g. MSI) or skewed towards early CRC stages (e.g. polyps). Interestingly, patients with LRAT hypermethylation did present a favorable prognosis (p = 0.03). Consistent with our finding, a recent study has also demonstrated high expression of LRAT in melanoma metastases was inversely correlated with patient survival [12]. Our data suggest that the frequent LRAT hypermethylation may represent a useful predictor to stratify CRC. Since LRAT promoter hypermethylation results in decreased gene expression, we hypothesize that reduced LRAT gene expression may involve a defending or counteracting mechanism to CRC tumorigenesis.
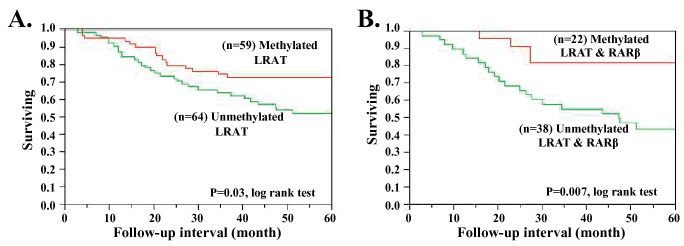 Figure 1: Kaplan-Meier survival analysis of CRC tissues with (A) LRAT hypermethylation status; (B) LRAT and RARβ combined hypermethylation status.
View Figure 1
Figure 1: Kaplan-Meier survival analysis of CRC tissues with (A) LRAT hypermethylation status; (B) LRAT and RARβ combined hypermethylation status.
View Figure 1
To further investigate the impact of reduced LRAT expression on the growth of CRC cells, DLD1 and HT29 cell lines were transfected with LRAT small interfering RNA (siRNA) which was designed to target the human LRAT sequence (NM_004744) position 3272-3296, with the sense targeting sequence: CAAGGAGGGAGGAU-CACAAGGUCAG. A duplex Dicer-substrate siRNA with a scrambled sequence (DS ScrambledNeg) that does not target any sequence in the human genome was used as a universal negative control. For each condition, 3 × 103 CRC cells were seeded in each well of the 96-well plates on day one and assayed in triplicates. A final 10 nM siRNAs were transfected into DLD1 and HT29 cells using RNAiMax (Invitrogen) under the manufacturer recommended condition, in the presence or absence of retinol. LRAT RNAs were measured 48 hours after transfections using qRT-PCR to determine the transfection and knockdown efficiencies. Cells were harvested every 24 hours after the first two-day of transfection and subjected to a MTT cell proliferation assay (ATCC).
As shown in Figure 2, in the absence (0 μM) of retinol, there was no difference in cell proliferation between LRAT knockdown and control cases. Remarkably, in the presence of 1 μM retinol, LRAT knockdown cells showed slower growths than the controls. These results imply that reduced LRAT expression is linked to a slow proliferation of CRC cells mediated by retinol, and may underlie the association between LRAT methylation silencing and a better prognosis of CRC.
 Figure 2: siRNA targets LRAT in CRC cell lines. The optical density was measured at 450 nm for cell proliferation assay. Red and blue curves represent the presence or absence of LRAT interference treatment, respectively. Retinol concentration used for each assay is shown in the parenthesis next to the cell line name.
View Figure 2
Figure 2: siRNA targets LRAT in CRC cell lines. The optical density was measured at 450 nm for cell proliferation assay. Red and blue curves represent the presence or absence of LRAT interference treatment, respectively. Retinol concentration used for each assay is shown in the parenthesis next to the cell line name.
View Figure 2
An explanation to LRAT-involved slow cell proliferation may attribute to mechanisms of retinol mediated CRC growth inhibition. Reduced LRAT expression impairs the esterification of excess retinol. The unconverted retinol is then metabolized into retinoic acid and contributes to antiproliferation effects likely through a Retinoic Acid Receptor (RAR) pathway [13]. Alternatively, several publications have proposed another mechanism suggesting a RAR-independent pathway for retinol-mediated growth inhibition of CRC and melanoma cells [1,2,14]. Our data also suggest that LRAT methylation silencing facilitates the utilization of retinol in regulating cellular proliferation. Furthermore, our analysis of RAR methylation status has shown that CRC patients with promoter hypermethylation in both LRAT and RARβ correlated with a better prognosis (p = 0.007, Figure 1B). Although this finding does not preclude the participation of LRAT in the anti-proliferation mechanism involving retinoic acid and its receptors, the association between a good survival outcome and hypermethylation at both LRAT and RARβ loci suggests that methylation silenced LRAT may be a key feature in a RAR-independent pathway to tumorigenesis inhibition.
LRAT activity is governed by different mechanisms. Vitamin A and its main metabolite (retinoic acid) regulate both LRAT mRNA and gene activity in a tissue specific manner [15]. Study in animal model suggested a second mechanism of regulating LRAT expression via CRBP. The ratio of free CRBP to holo-CRBP directly reflects vitamin A concentration and influence its regulatory ability [16,17]. Here, we propose LRAT promoter hypermethylation as another mechanism for modulating its gene expression. It has been proposed that LRAT, in conjunction with cytochrome oxydases P450s, plays an essential role in regulating the availability of retinol as a precursor for retinoic acid [16]. Methylation silencing of LRAT may abolish the feedback regulation and disrupt the homeostasis of a careful balance between retinoic acid and retinol concentration. Consequently, such an imbalance may interfere with regular cell growth, differentiation and apoptosis. Future studies to investigate LRAT’s roles in each of the retinol mediated inhibitory mechanisms will shed light on this interesting topic.
Funding for this work was provided by the Clinical Nutrition Research Unit P30-CA29502 (YWC), National Cancer Institute P01-CA65930 (FB), Ludwig Institute for Cancer Research/Conrad N. Hilton Foundation joint Hilton-Ludwig Cancer Metastasis Initiative, and the Gilbert Family Foundation. The authors thank WeiJi Shi for the advice in statistical analysis. We also thank Owen Parker and Jianmin Huang for insightful discussion and critical reading of the manuscript.