Aerobic oxidation of carbohydrates by mitochondria has been a great invention of nature, marking a big step in early evolution. This biological system has greatly multiplied the energy production of the cell by supplying this energy to different metabolic units, with increased temporal and spatial specificities. The numerous mitochondrial functions that have emerged and developed along evolution constitute strong signatures of evolution pressures, driving toward higher order, more complex and more energy-expending organisms. In evolution, there is a firm tendency to generate complexity, with larger DNA-containing systems. Therefore it is obvious that these progresses in evolution require an increased in energy production with a better cellular distribution. An overview of few representative mitochondrial functions and a look inside gene evolution of particular examples is a good way to approach and emphasize this process. In order to insist on how mitochondria can guarantee both life and evolution, we have focused on a few mitochondria-linked functions and their phylogenies, such as respiration per se, ATP/ADP and solute transports, contact sites and biogenesis systems, mitochondrial fission/fusion and distribution, and calcium as a central regulator of mitochondrial functions. We will also shed light on the genetic evolution of a very recently discovered mitochondrial protein called ATAD3 that evokes perfectly how evolution has improved discretely but efficiently mitochondrial capacities.
To become larger and stronger organism need to expend energy. Phosphorylating oxidation is probably the best complex bioenergetic process to achieve this goal. Indeed, the genetic basis of mitochondrial functions has greatly evolved across species since the original endosymbionte [1-4]. Even if less genes encumber mitochondria during evolution, as the original genes were transferred to the nucleus, the overall genetic functions of mitochondria have greatly evolved, to encode a mitoproteome of at least 1500 proteins in human for example [5-9].
If the number of mitochondrial functions has increased enormously, a main goal of evolution has been to target mitochondria that is to send and move these energetic factories to areas where energy is required. Also, of course, mitochondria are biological entities, so they live, function, renew or die. In addition, a major function of mitochondria that has appeared as an important factor for life is thermogenesis, which is necessary for regular gene expressions and enzymatic activities.
In all cases, the "vicious circle" of mitochondrial evolution is that more functions and more genes always require more energy, in order to preserve and express these genes. This can be seen by the tight correlation between the metabolic rate and the body mass observed in all species inside unicellular organisms, poikilotherms and homeotherms. In the same way, the respiratory capacities are clearly and positively correlated with the size of organisms, as observed by the tight correlation between the respiratory apparatus surface and the body weight in fishes and mammalians. Then, as animals became denser and heavier, walking became the only way for the most evolved species to move. In contrast, in flying and swimming animals, where less energy is consumed for movement, the density of body was never allowed to increase as much as walking has permitted.
There are two major landmarks in mitochondria evolution: the increase in functionalities and efficiency, and the incredible transfer of mitochondrial genes to the nucleus.
First, let us consider the numerous functions and regulation that are now associated with and assumed by mitochondria. Mitochondria obviously need to organize and optimize their own metabolism. The evolution improved ATP/ADP transport across mitochondrial membranes, which is the basic mitochondrial function of providing ATP to the cell. But evolution improved also all the mechanisms providing mitochondria with metals, amino-acids, RNAs and others such nucleotides that are all necessary for mitochondrial transcription, translation and enzyme activities. Mitochondria were also provided with lipids to ensure integrity and biogenesis of the membrane systems. Furthermore these functionalities have evolved across all species.
Moreover, the mitochondrial mass is regulated, for example at birth and during growth or aging, as well as following training or diets. These regulations depend on different factors such as nutrition, seasons, or state of health. Therefore, mitochondria need to be regulated in size and number, as well as to be renewed or eliminated (autophagy). For this, they now exist in higher eukaryotes a complex set of regulations that able efficient mitochondrial biogenesis and destruction. These processes are tightly linked to the endoplasmic reticulum and other compartments which provide mitochondria specially with lipids [10] but also with all necessary molecules like nuclear-encoded 5S rRNA or tRNA [11] and with of course all the translational products of nuclear mRNAs encoding mitochondrial proteins [12,13]. Today all these mitochondrial biogenesis processes are better understood and research shows that their functions and regulations have greatly evolved [14].
Having maintained and regulated their mitochondrial mass, cells then benefited from further improvements in mitochondrial functions such as mitochondria targeting to specific places where energy is required. For this purpose, mitochondria need to be produced, divided, moved away by motors and docked at their assigned places. All these processes require specific molecular signatures because their movements to docking places are originally and specifically defined. These so-called "mitochondria-containing micro-domains" are now better known and among them, the best example is the neuronal synapse [15].
These last evolutionary progresses in mitochondrial functions have probably led to a large improvement in the energy use and cellular capacities. Furthermore, bioenergetic evolution ultimately allowed for precise regulations of mitochondrial activities and mass as regard to cell destiny.
Secondly and interestingly, mitochondrial DNA (mtDNA) has greatly decreased during evolution, in contrast with the functional increase [3,16]. How this has occurred across evolution still remains today poorly understood. Possible explanations could be that nuclear genes were pre-existing as double copies, or that gene transfer occurred by phage/virus, or even that other processes linked to reverse-transcription could have moved mtRNA coding sequences to the nucleus.
As a consequence, regardless of the mechanism implicated, nuclear genome evolved toward a better management of mitochondrial functions in general [17].
In this short review, we will briefly describe some of the particular mitochondrial processes and functions, pointing out major evolution advances and their timing, insisting mostly on mitochondrial functions that have been improved to produce energy when and where needed, as cells are always aiming for greater efficiency. We will also underline, when known, how mitochondrial dysfunctions can cause diseases.
OXPHOS: OXPHOS proteins comprise over 86 subunits in human (CI is 45 subunits, CII is 4, CIII is 11, CIV is 13 and CV is 13) and represent the best example to describe mitochondria evolution history [18]. Indeed, the number of subunits has greatly increased from bacteria to human, and consequently their regulation pathways has become more complex.
The phosphorylative oxidation is the central function of mitochondria and the corresponding genes are encoded both by the nucleus and the mtDNA, and therefore their expressions are necessarily very tightly coupled [19]. As an example, we can just focus on the F0/F1 ATP-synthase (complex V) that produces ATP from ADP using the proton-motor force generated by the respiratory chain. This complex is a powerful window through which evolution can be observed.
As example two subunits are described below, one nuclear-encoded, ATP5G1, and a second mitochondria-encoded, MT-ATP6.
ATP5G1: ATP5G1 is the subunit 9 of the H+-transporting mitochondrial F0 complex subunit C1 of the F-type ATPase. F-type ATPases are composed of two structural domains, the F1, which contains the extra-membraneous catalytic core, and the F0, which contains the membrane H+ channel. They are linked together by forming the central and peripheral stalks. Catalysis of ATP synthesis, in the F1-catalytic domain, is associated with a rotary process of the central subunits that translocate protons. ATP5G1 is part of the F0-complex which is a homomeric c-ring of 10 subunits involved in the rotary complex system. Two major known diseases linked to ATP5G1 dysfunctions are the "spindle cell hemagioma" and one form of coronary heart disease.
There are three nuclear genes encoding ATP5 subunits under three different pre-imported isoforms to produce finally the same mature protein. ATP5G1 gene contains 5 exons and is located on chromosome 17 in human. It is transcribed for the production of 10 differentially-spliced transcripts. Even with some minor variations, the protein sequence is highly conserved among species and is present only from the common ancestors of animal and fungi (see phylogenic tree Figure 1). There are also 9 pseudogenes (with no promoters) for ATP5G1.
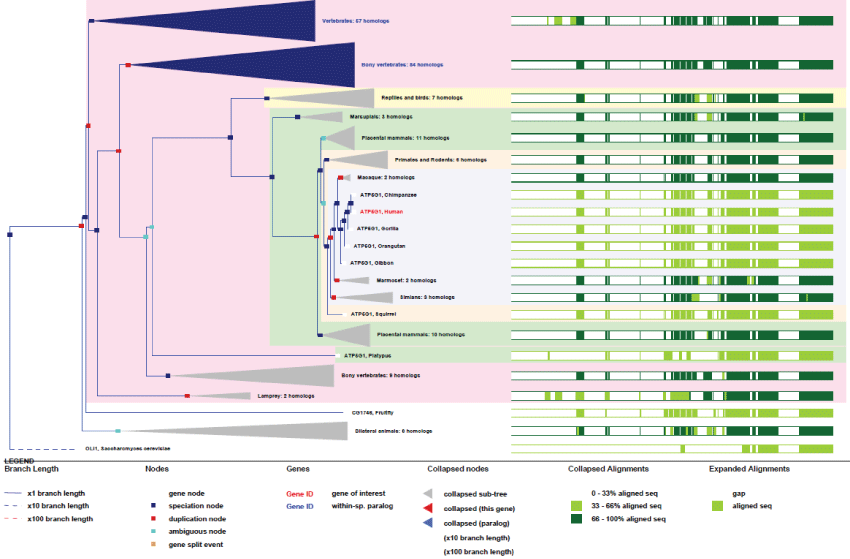 Figure 1: Phylogenic tree of ATP5G1. View Figure 1
Figure 1: Phylogenic tree of ATP5G1. View Figure 1
As it can be seen in figure 1, the ATP5G1 gene, even with few duplications, remained very conserved, without particular improvement in its basic function, excepted for few regulation sites that appeared from differentially-spliced transcripts. An important point to be noted about F1-ATPase is that evolution brought to homeothermes the uncoupling action of UCPs proteins that abled the shutdown of F1-ATPase activity while creating heat from the chemical-gradient proto-motor energy [20].
MT-ATP6: MT-ATP6 is the main subunit of the proton channel and is playing a direct role in the protons translocation driven by the whole ATP synthase activity.
Like all mitochondrial genes, ATP6 gene does not contain any exon. First appeared in yeast, it remained highly conserved (see phylogenic tree Figure 2). It probably corresponds, like few other mtDNA genes, to one of original genes involved in the first events of endosymbiosis.
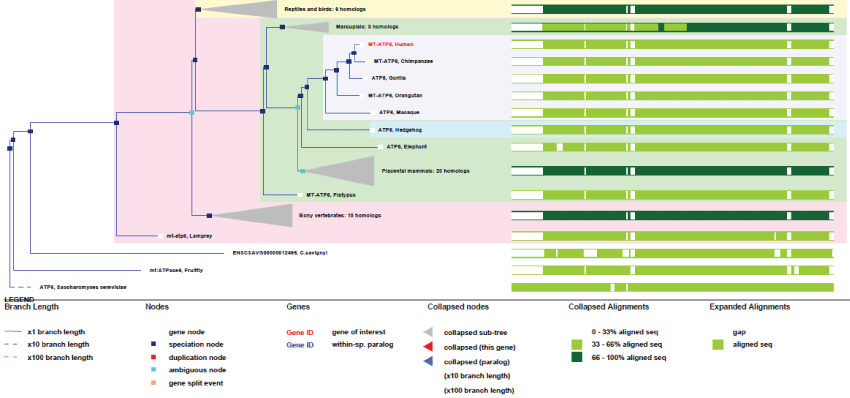 Figure 2: Phylogenic tree of MT-ATP6.View Figure 2
Figure 2: Phylogenic tree of MT-ATP6.View Figure 2
As we can see in figure 2, MT-ATP6 remained with no duplication and very high conservation of homology. Therefore, no specific improvement of this gene appeared during evolution, indicating how high the selection pressure on this particular function has been. Of course, we may not expect gene-duplication in mtDNA, as the tendency of evolution is inexorably to decrease mtDNA size, but one must admit that this gene, like other remained mitochondrial genes, might not have yet been transferred to the nucleus.
These two first examples of mitochondrial proteins and their corresponding genes, as different as they are, show how much evolutionary distance exists for such a tightly coupled system, where the concerted expression of these subunits happens in both nuclear and mitochondrial compartments. MtDNA encoded genes are typical ancestral prokaryotic genes, intron-less, without variants but with a strongly coupled transcription/translation. On the opposite, nuclear-encoded genes are archetypes of more recent and highly evolved genes. In general they stay as "unique genes", but containing many exons that allow for production of many transcripts that can generate specific tissue- or developmental isoforms.
When describing the basic mechanism of respiration driven by proton exchange and ATP synthesis, one must consider other related mitochondrial functions.
In order for mitochondria to be accepted as a symbionte, one of the first and oldest system is the export of ATP and its renewal from ADP imported back into mitochondria. This essential process involves Adenosine Nucleotide Translocase (ANT) and Voltage-Dependent Anion Channel (VDAC) [21-23]. ANT is the inner membrane ATP/ADP specific carrier while VDAC is the outer membrane non-specific transporter.
ANT4 (SLC25A31): ANT catalyzes the exchange of ADP from the inter-membrane space with ATP from the matrix across the mitochondrial inner membrane. The function of ANT is of course vital and its structure and functions have been well studied. Furthermore, mutations of ANT have been shown to be involved in severe cardio-myopathies like in skeletal myopathies and some neuropathies.
A remarkable and interesting feature of ANT, like for other mitochondrial genes, is that many pseudogenes exist in human. We may suppose that pseudogenes might guarantee over time the survival of the original gene. As an example, ANT4 is one of four ANT genes that differ very little from one another. This gene, located on chromosome 4 contains 4 exons and is expressed under 2 spliced variants. The gene exists in yeast and was present in the common ancestor of eukaryotes. It has remained very conserved during evolution (Figure 3).
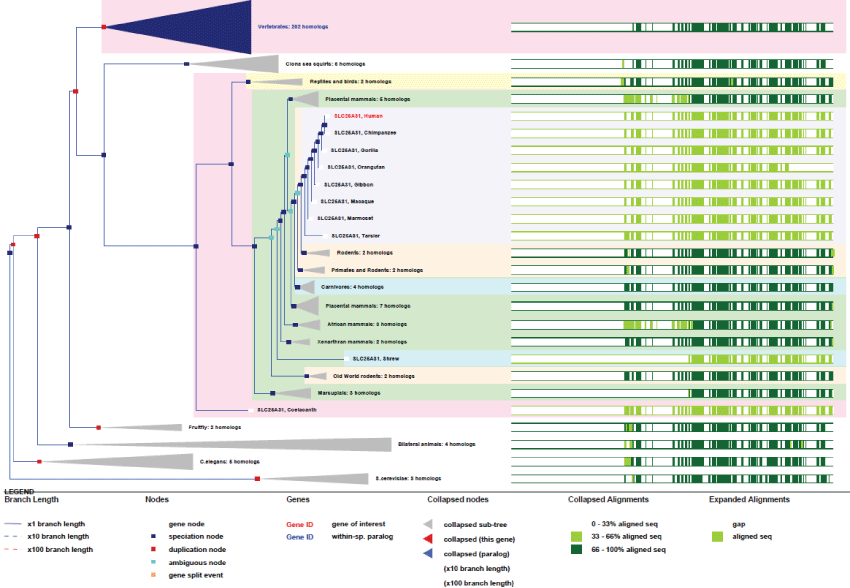 Figure 3: Phylogenic tree of ANT4.View Figure 3
Figure 3: Phylogenic tree of ANT4.View Figure 3
As we can see in figure 3, the only improvements observed for the well conserved ANT4 concern the existence of only 2 differentially spliced mRNA variants. Also, duplications of the gene occurred very rarely and quite early on. As mentioned before, the 3 other genes for ANT, ANT1, ANT2 and ANT3 are located on chromosome X and present the same genetic characteristics. Here again, it seems clear that this gene is maintained by a very strong evolution pressure.
VDAC1: VDAC belongs to the porin family and forms channels across the mitochondrial outer membrane as well as across the plasma membrane. Outer mitochondrial membrane VDAC permits diffusion of hydrophilic molecules of small sizes (less than 10 kDa). At the plasma membrane, VDAC is implicated in cell volume regulation and apoptosis. VDAC can assume an open conformation at low membrane potential and a closed conformation at potentials above 40 mV. While the open state presents a low selectivity for anions, the closed state is selective for cations.
Like for ANT, VDAC gene presents many pseudogenes. Its location is on chromosome 5 and it contains 8 exons and 7 splicing mRNA variants. The gene is very conserved across evolution and is present in the common ancestor of animals and fungi (see phylogenic tree Figure 4).
 Figure 4: Phylogenic tree of VDAC1.View Figure 4
Figure 4: Phylogenic tree of VDAC1.View Figure 4
As it can be seen on figure 4, VDAC appeared early in yeast and the gene evolved by several duplication, while the function and the specific structural features remained highly conserved. Also there exist two other VDAC genes, VDAC2 and VDAC3, present on chromosome 10 and 8 respectively. They consist of 10 exons and 14 spliced variants. Like other ancient genes, such as ATP5G, the high level of homology in VDAC strongly demonstrates the importance of the evolution pressure on this channelling process.
Of course, many other transporters are necessary for all the mitochondrial functions that require import of molecules such as metals, amino-acids, lipids and RNA like tRNA and 5S rRNA [24].
For more than 10 years, the dynamic characteristics of mitochondria have been well investigated. To proliferate and disperse within the cell, mitochondria need to import many cellular metabolites. Different biosynthetic fluxes occur and several of them originate from the endoplasmic reticulum. These transport mechanisms provide mitochondria with lipids and proteins, and today new ideas are emerging, like the concept of Mitochondria Associated Membranes MAM, [14] or other kind of lipid raft-based transfer processes [10] to explain how these molecules can reach the different mitochondrial compartments.
Many proteins are involved in these contact sites and translocation systems and theses interactions tightly control mitochondrial DNA synthesis [25]. We will now focus on two of these proteins, namely ATAD3, that is functionally linking the reticulum to the inner mitochondrial membrane and the other one, OPA1, linking the outer and inner mitochondrial membranes to the matrix systems.
ATAD3: ATPase family AAA Domain-containing protein 3 (ATAD3) is a newly described ATPase located in the inner mitochondrial membrane. Although its topology is today quite well understood [26-29] but the precise function of the protein is still unknown despite being linked to the nucleoid organization-activity [30]. Importantly, the ATAD3 knock-down is lethal for Caenorhabditis elegans, Drosophila melanogaster and mouse early developments, thus indicating that it plays a major role in cell growth and differentiation. All investigations have led today to the idea that this gene is involved in mitochondrial biogenesis. At first, ATAD3 has been described as the target gene of c-Myc. Thereafter, it has been found overexpressed in several cancers and shown at the same time to be a potential inhibitor of cell cycle and of apoptosis, as well as a chemoresistance conferring gene (for review see Li and Rousseau 2012 [29]). Precise localization and topology of the protein were then established by different approaches leading to the model where the canonical C-terminal ATPase core domain of ATAD3 is located into the mitochondrial matrix and that the N-terminal domain is either in the inter-membrane space, or the outer membrane, or in the cytoplasm. Moreover, ATAD3 homo-polymerizes like many other ATPases but specifically interacts with protein S100B, a PKC-regulator. Experiments with si/shRNA in primary and immortalized cultured cells have showed that ATAD3 is essential for the maintenance of the mitochondrial network [31] as well as well as for the reticulum/mitochondria interactions such as those involved in cholesterol transfer between these compartments [32]. However, the functions associated with ATAD3 ATPase domain still remain to be elucidated. The identification of PKC phosphorylation sites and of a S100B binding domain on ATAD3 confirmed that this protein is involved in cell growth processes including pathological conditions like cancer progression. Although, many studies were focused on the major ATAD3 protein variant, p66 (66-68 kDa in mammals), several proteins and RNA isoforms exist in rodents and humans [33]. Accordingly, the pattern of ATAD3 expression appears much less simple than initially assumed. At least five isoforms exist in humans, and display tissue-specific expressions. These isoforms are generated by both differential splicing and differential translation initiation mechanisms.
ATAD3 exists as 3 genes in human (one gene in rodents) that are localized adjacent to each other in the distal portion of chromosome 1. These genes all contain many exons (13 for ATAD3A) and produce many transcripts. As it is not present in yeast, the unique ancestral gene likely appeared in pluri-cellular organisms. It is noteworthy to point out that this unique gene has duplicated twice from rodents to primates where all 3 genes are juxtaposed [33]. Also, contrary to what one might expected from its central function, no disease is actually known to be linked to ATAD3 dysfunction. Nonetheless, the essential function of ATAD3 in controlling nucleoid structure and function, like cholesterol import, could represent a possible origin of mitochondria-associated diseases [34].
As we can see on figures 5 and figure 6, ATAD3 genes duplicated only twice and very recently in primates showing how much ATAD3 expression levels need to be controlled. Like for the other genes cited prior, with the exception of MT-ATP6, all these genes have evolved and increase their functionalities by creating isoforms through alternative transcription, splicing, translation and new post-translational regulations. Here again as a significant example, as recent studies on ATAD3B have determined new phosphorylation sites on the C-terminus of the protein, as well as a Nuclear Export Signal (NES) that indicates new and interesting functionalities for a mitochondrial protein.
 Figure 5: Phylogenic tree of ATAD3A.View Figure 5
Figure 5: Phylogenic tree of ATAD3A.View Figure 5
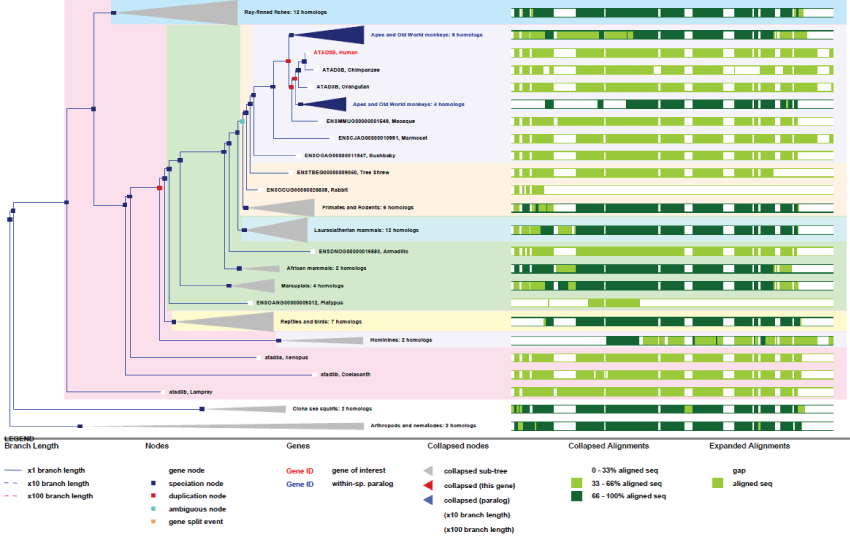 Figure 6: Phylogenic tree of ATAD3B.View Figure 6
Figure 6: Phylogenic tree of ATAD3B.View Figure 6
The example of ATAD3 genes interestingly marks both early and recent evolution steps, like the ancestral gene remaining from first multicellular organisms and the appearance of new genes specific to primates. However, the fact that the protein is ubiquitous, expressed in all organs, lead us to think that the related function is not specific to certain organs like brain or linked to communication behaviours in primates. Obviously it is present in all organs, so it might associated with improvement of mitochondrial biogenesis pathways and remodelling or targeting in general.
OPA1: Optic Atrophy protein 1 (OPA1) is a so called "dynamin-related" GTPase which is required for mitochondrial fusion. OPA1 protein is involved in several functions such as stabilizing the respiratory chain and controlling the membrane potential and cristae organization. It is also involved in apoptosis and in mitochondrial DNA integrity. OPA1 is located in the mitochondrial inner membrane but can also interact with MFN1 and MFN2 at the mitochondrial outer membrane. Therefore, its role may be principally linked to mitochondrial fusion regulations.
OPA1 is also an essential protein and associated mutations have severe health impacts. OPA1 mutation is linked to progressive visual loss and in parallel to optical atrophy. These mutations affect a number of neuronal structures in the retina, optic disk, nerve and chiasm. OPA1 is also involved in apoptotic-linked processes [35].
OPA1 human gene is unique and found chromosome 3. It contains an impressive number of exons (24) and is expressed under 19 transcribed forms. The gene appeared early in the ancestor of animals and fungi and remained very conserved.
As it can be seen in figure 7, OPA1 gene duplicated only twice along evolution, and its conservation degree is very high, more than ATAD3. As revealed by the high conservation of genes and of their related functions, as well as their rare or inexistent duplication, evolution did not need many changes in the principal mitochondrial functions involved in contact sites and biogenesis of mitochondrial fluxes. This imply that the Krebs cycle was already well developed from yeast, meaning that the "perfection" of the system was achieved early by eukaryotic cells; all this reflecting also strong evolution pressures. In contrast, as illustrated by ATAD3 genes, some mitochondrial functions have evolved more recently and improved in primates by gene duplication. Also, other unique genes like OPA1 evolved by producing many new variants through differential splicing and translation initiation.
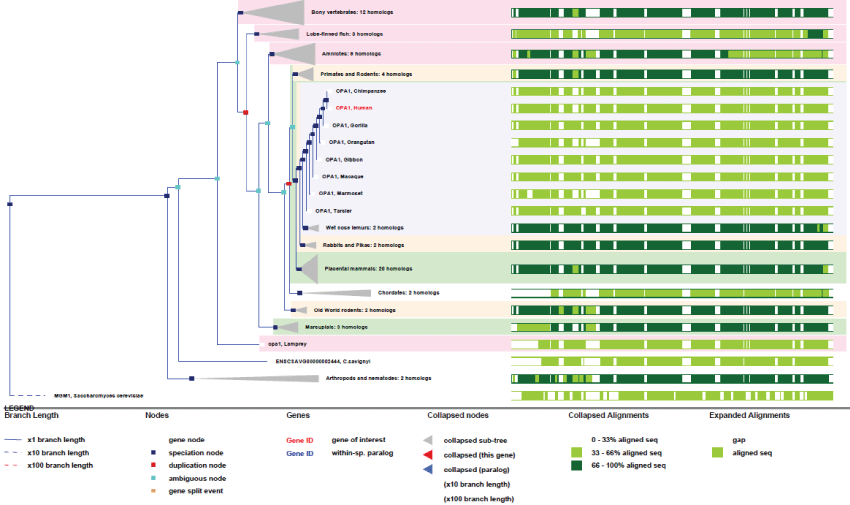 Figure 7: Phylogenic tree of OPA1.View Figure 7
Figure 7: Phylogenic tree of OPA1.View Figure 7
If mitochondrial biogenesis can lead to an increase in mitochondrial mass and biological functions, it is clear that fission and displacement events have to occur in order for the mitochondria to multiply and position at expected sites, especially during proliferation and differentiation. This is obviously observed at ultra-structural level, in all organs like muscle, brain, kidney and spermatozoids or ovules. Theses fission events occur first through the action of DRP1/MFN2 fission system [36] and the transport of mitochondria is then ensured by specific motors like kinesin, involving cytoskeleton structures as transport paths [37]. Then, finally, a docking system is required at the right place involving other structures like intermediate filaments.
It is clear that mitochondria have to acquire a molecular phenotype, a proteomic signature, in order to be moved and docked at their right places. The molecular specificity appears early in the biogenesis of mitochondria. This is of special interest because it implies necessarily an anticipated regulation of nuclear and mitochondrial genes expression for what may corresponds to the differentiation program.
Since cellular differentiation is very diversified from yeast to human it is evident that these processes have however greatly evolved and that they are most probably linked to the improvement of mitochondria targeting.
DRP1: Dynamin-related protein 1 (DRP1) operates during mitochondrial and peroxisomal divisions by inducing membrane fission [38]. Fusion/fission of mitochondria proceed in most cell types and constitutes an important step in the regulation of mitochondrial network structure, balanced between fusion and fission. DRP1 oligomerizes to form membrane-associated tubular structures that constrict around the scission site by a GTP-dependent hydrolysis process. Because of its involvement in mitochondrial division, DRP1 participates in most mitochondrial regulations. It is required in processes such as normal cytochrome c release and caspase activation upon apoptosis. DRP1 is also involved in mitochondrial fission during mitosis, participates in the formation of endocytic vesicles and regulates synaptic vesicle membrane dynamics. DRP1 is required for normal brain development because mutations has been linked to encephalopathies, caused by defective mitochondrial and peroxisomal fission. These autosomal dominant diseases are characterized with defects in neurologic development and early death.
DRP1 human gene is unique and found on chromosome 12. It includes 17 exons and is expressed under 8 isoforms arising from 30 splicing mRNA variants. This well conserved gene is already present in yeast and in the common ancestor of eukaryotes.
As we can see in figure 8, DRP1 is an ancient or initial mitochondria-associated protein and its functional domains remained tightly conserved across evolution. Again, the interplay of differential splicing and translation allow one gene to produce a large number of developmental and tissue-specific isoforms.
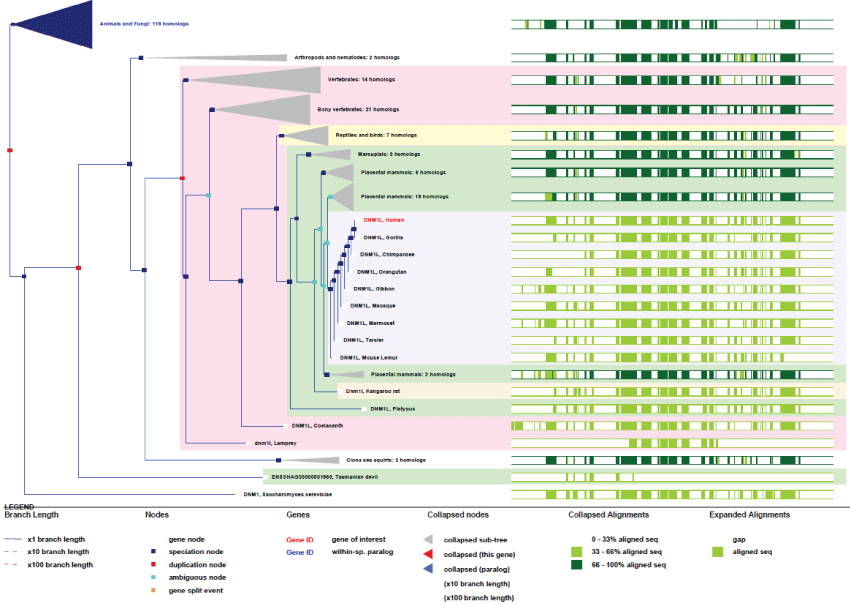 Figure 8: Phylogenic tree of DRP1.View Figure 8
Figure 8: Phylogenic tree of DRP1.View Figure 8
MFN2: Mitofusin 2 (MFN2), is an essential transmembrane GTPase mediating mitochondrial fusion, located in both the outer mitochondrial membrane and the endoplasmic reticulum membrane. MFN2 plays a major role in mitochondrial metabolism and its deregulated expression is associated with obesity and apoptosis. MFN2 expression regulates the formation of the mitochondrial network and thus is important in the cell cycle regulation. It is also involved in endoplasmic reticulum stress responses and mitophagy [39].
MFN2 mutations are known from the Charcot-Marie-Tooth disease 2A2, an axonal form of the disease. Charcot-Marie-Tooth diseases are peripheral nervous system disorders characterized by progressive weakness and atrophy. These diseases are classified depending on electrophysiological properties, nerve conduction velocities, primary peripheral demyelinization, axonal degeneration, distal muscle weakness and atrophy. MFN2 deficiency is also associated with severe early-onset axonal neuropathy and hereditary motor and sensory neuropathy type 5/6.
MFN2 human gene is on chromosome 1 and composed of 17 exons which produces 6 transcripts. The gene does not exist in yeast but appeared in worms and remained very conserved thereafter. There exists also another MFN gene, MFN1, which is on chromosome 3, composed of 18 exons and producing 9 transcripts.
As can be seen in figure 9, although many splicing variants have appeared during evolution together with new regulating functions (phosphorylation sites, tissue and development specificities), the MFN2 gene stayed very highly conserved among all species, pointing out again how much these functions are essential for life. The MFN2 gene is present in the common ancestor of animals but not in yeast. It is probable that MFN1 is the ancestor gene. It is important to note that, because MFN2 (like ATAD3) appeared with pluri-cellular organization, MFN2-related protein functions may be directly linked to the appearance of cell differentiation and structural organization within cells.
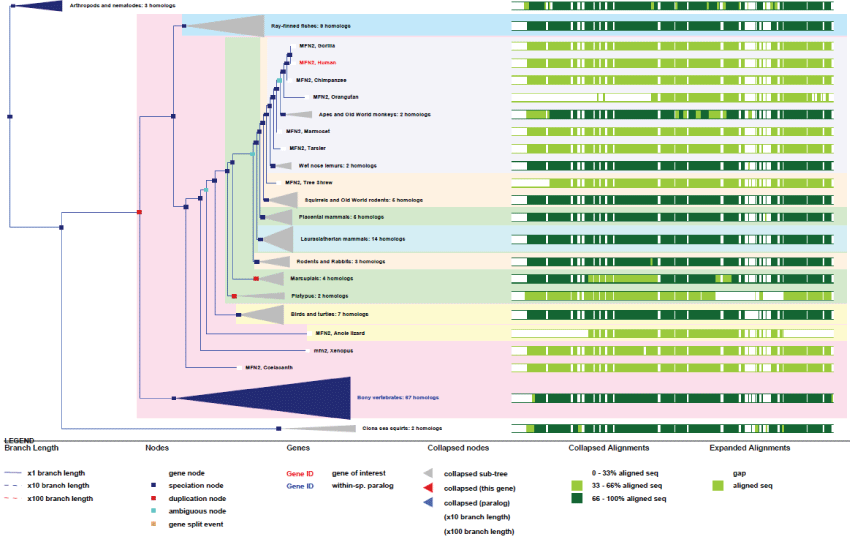 Figure 9: Phylogenic tree of MFN2.View Figure 9
Figure 9: Phylogenic tree of MFN2.View Figure 9
DRP1 and MFN2 are therefore major actors of mitochondrial fusion/fission processes, and also of differentiation. However, movement and targeting depend of motor proteins, like kinesin, and proteins of the cytoskeleton. These molecules multiplied and diversified along evolution and surely contributed to the improvement of mitochondria addressing to different cell type structures.
The role of mitochondria in lipids metabolism is today quite well understood, even if several questions remain open. The implication of mitochondria in lipids metabolism is an important evolution feature [40-44]. A good example of this is the contribution of mitochondria to myelinisation of axons by oligodendrocytes. These cells produce sphingomyelin through mitochondrial steps, starting from phosphatidyl-serine (PS) to make phosphatidyl-ethanolamine (PE) and then phosphatidyl-choline (PC) produced at the inner mitochondrial membrane (IMM). PS is primarily transferred from the endoplasmic reticulum reservoir to the IMM and then decarboxylated by the Phosphatidyl-Serine Decarboxylase (PISD) to be further processed into PC and sphingomyelin [45,46]. The synthesis of sphingomyelin is performed by the enzymatic transfer of phospho-choline from PC to ceramide, with initial steps involving the condensation of L-serine and of palmitoyl-CoA catalyzed by the serine palmitoyl-transferase. Sphingomyelin synthesis reactions occur at the cytosolic face of the endoplasmic reticulum. Ceramides are transported to the Golgi apparatus and converted to sphingomyelin by the sphingomyelin synthase.
Mitochondria are involved also in many other lipid processes like cholesterol-steroid synthesis, β-oxydation of lipids and others. Among many, I will focus on two specific protein involved in particular mitochondrial lipid processes.
PISD: Phosphatidyl-Serine Decarboxylase (PISD) is a protein bound to the inner mitochondrial membrane, at the matrix side. PISD catalyses the conversion of PS into PE, producing CO2. Phosphatidyl-ethanolamine is further transformed into different cellular lipids like PC [40].
The human PSID gene is unique, located on chromosome 22 and composed of 9 exons. The gene is expressed under 17 transcripts and exists in the yeast as in the common ancestor of eukaryotes.
As it can be seen in figure 10, the gene structure of PISD remained very conserved, indicating a strong selection pressure on these mitochondrial functions. A second isoform of PISD, with a modified Nter-sequence also exists as a result of differential splicing and translation initiation.
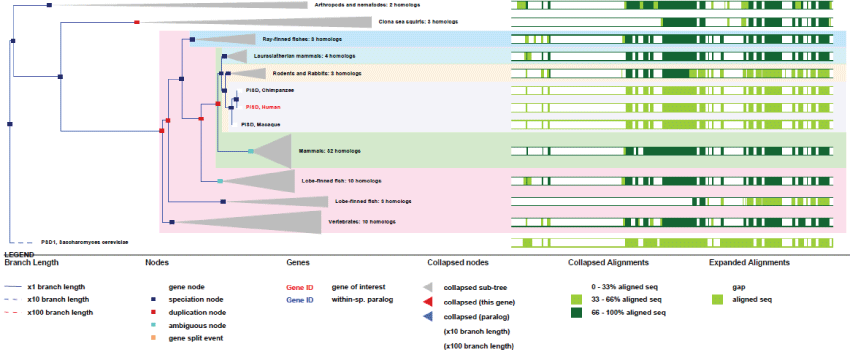 Figure 10: Phylogenic tree of PISD.View Figure 10
Figure 10: Phylogenic tree of PISD.View Figure 10
Cardiolipin (CL) is another important lipid for mitochondria. It is almost mostly exclusively present in the IMM inner leaflet and produced by mitochondria through several steps, some of which still remained unknown [10].
The mitochondrial synthesis of CL uses phosphatidic acid (PA) as precursor. PA has to be imported from the ER, probably by contact sites. The produced CL can then be exported out the mitochondria to be transformed in the ER. In contrast to OMM and to other cellular membranes that are virtually devoid of this phospholipid, CL is produced and highly enriched in IMM. Therefore, this heterogeneity is probably responsible in part for the mitochondrial shape/structure and for the specific IMM-distributed functions.
CRLS1: The cardiolipin synthase 1 (CRLS1) is involved in catalyzing the reversible transfer of the phosphatidyl group from one phosphatidyl-glycerol molecule to another one in order to form CL (diphosphatidylglycerol) and glycerol [10]. Its deficit is linked to diseases such as the catastrophic anti-phospholipid syndrome.
CRLS1 is a unique gene present on chromosome 20. It has 7 exons and gives rise to 7 transcripts. The gene exists in yeast and in the common ancestors of animals and fungi. Two isoforms can be produced, one with a N-terminal deletion.
As it can be seen in figure 11, an early ancient mitochondrial gene, CRLS1 is surely and initially essentially for mitochondria, as shown by its high degree of conservation. Also, as a point of interest, CRLS1 was never duplicated during evolution.
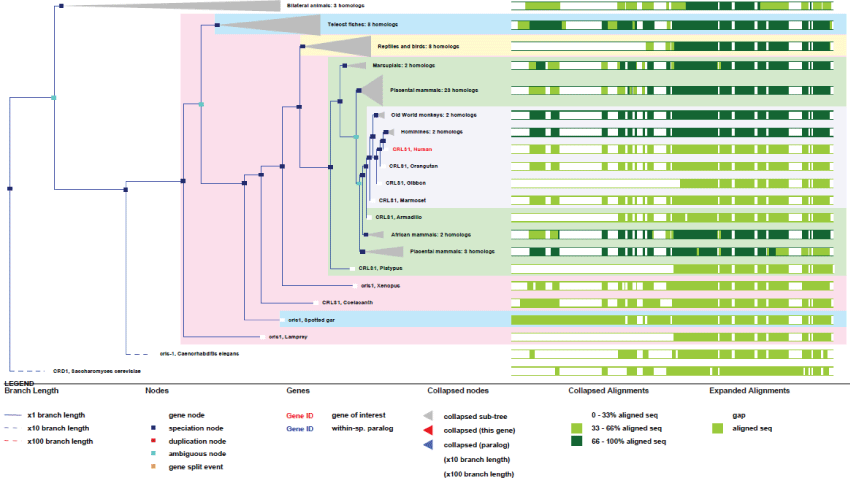 Figure 11: Phylogenic tree of CRLS1.View Figure 11
Figure 11: Phylogenic tree of CRLS1.View Figure 11
Again, these two typical genes involved in lipid metabolism have stayed unique and very conserved, producing only variants by differential splicing.
Another aspect about mitochondria and lipids metabolism is the case of steroids that are produced in mitochondria through modifications of cholesterol. Cholesterol is transported from the ER to the mitochondria and then processed. This transfer again happens at contacts sites between endoplasmic reticulum and mitochondria [32].
The molecular and genetic basis of all these mitochondrial lipid processes are still not completely understood and constitute new fields of research. Furthermore they may be related to unclassified mitochondrial diseases like myopathies and neuropathies.
Mitochondria have the particular ability to decode and transduce cytosolic signals like calcium into an energy output (ATP) according to the cellular demand. These regulations have surely been improved along evolution in order to ensure efficient energy usage. Specifically, mitochondrial calcium uptake, which occur as waves or sparks, modulates the cell bioenergy by increasing both the supply of reducing equivalents (to the electron transport chain, NADH production) and the F1-F0 ATP synthase activity (NADH consumption) [47]. Calcium also signals to mitochondrial functions through calci-proteins such as S100 and calmodulin together with phosphorylation signal transduction.
All these regulations contribute to the coordination of energy use and storage, as it is necessary for short term respiration modulation and also for mid and long-term adaptation, specially all hormone-based regulations. In addition, energy use needs to be adapted to the cell cycle, essentially at the beginning of the cycle, and also during differentiation [48]. Moreover this is true for variation of metabolism level during aging, stress responses or nutritional variations. Necessarily, all these adaptations need a tightly coupled signal transduction that reaches both the nuclear and mitochondrial compartments. Thus the recent discoveries of proteins that are able to shuttle between these two compartments does not come as a surprise.
Largen: Largen (PRR16) is a newly discovered protein which is involved in the translation of nuclear-encoded mitochondrial mRNAs [13]. The main characteristic of this protein is that its expression regulates cell size in mammals and promotes mitochondrial mass and respiration, in a mTOR and Hippo-independent manner [13]. Its specific mitochondrial location remains to be elucidated.
PRR16 is a unique gene present on chromosome 5. It contains 2 exons and produces 5 transcripts. The gene is not present in yeast nor in drosophila as it appeared late in higher eukaryotes. It is present in the common ancestor of chordates.
As it can be observed in figure 12 Largen gene appeared very late in fishes, never duplicated, and stays highly conserved in all species.
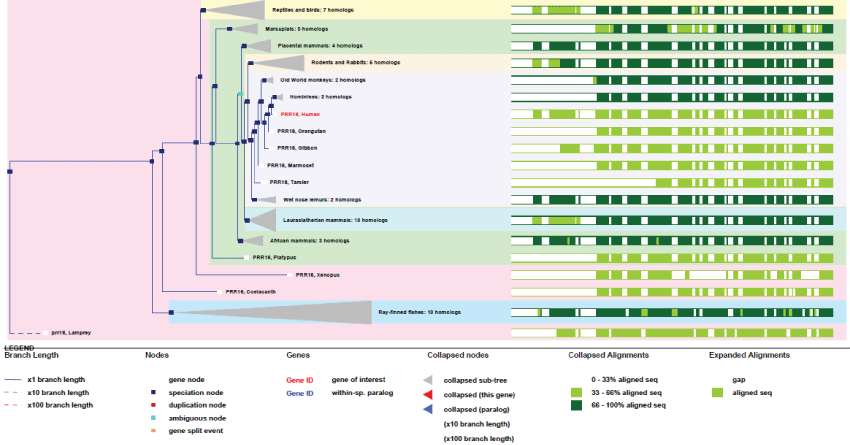 Figure 12: Phylogenic tree of Largen.View Figure 12
Figure 12: Phylogenic tree of Largen.View Figure 12
This gene, and its encoded protein, are of great interest because they represent an important landmark in late steps of evolution. As Largen, this new genes are able to increase and regulate cell size for all related purposes such as adaptation, thus creating a "cell size control". This involved a huge and still unknown field of nuclear-encoded mRNA translation and all pathways for the many mitochondrial proteins that need to be translocated and inserted in membranes.
S100B: The S100B is a protein sensor that binds calcium and zinc. It contains distinct binding sites with different affinities for each ions. Physiological concentrations of potassium can counteract both divalent cations binding and specially affects high-affinity calcium sites. S100B binds to STK38 (Serine/Threonine Kinase 38) to induce the activation of this kinase but also to PKC (Protein Kinase C), here for its inactivation. S100B is involved in myocardial infarction as playing a role in apoptosis by activating ERK1/2 and p53/TP53. S100B also regulates ATAD3 function and mediates calcium-dependent regulation of many physiological processes by interactions with other proteins, such as TPR-domain containing proteins. As the gene is located on chromosome 21, it is suspected to be the cause of some organs dysfunctions in Down syndrome [29] and also in Alzheimer disease, epilepsy, amyotrophic lateral sclerosis, melanoma and type I diabetes.
S100B gene contains 4 possible exons and produces 3 transcripts. The gene is not present in yeast and drosophila.
As it can be observed in figure 13, Like Largen, S100B appeared very late, in lamprey and fishes, but many different S100 genes exist in humans (19 genes). They are expressed in a tissue-specific manner and with different and specific range of partner targets [49]. S100B may therefore plays very important roles in metabolic activity regulation. Even if not yet well demonstrated, it is also believe that S100B might regulate mitochondrial mass [50].
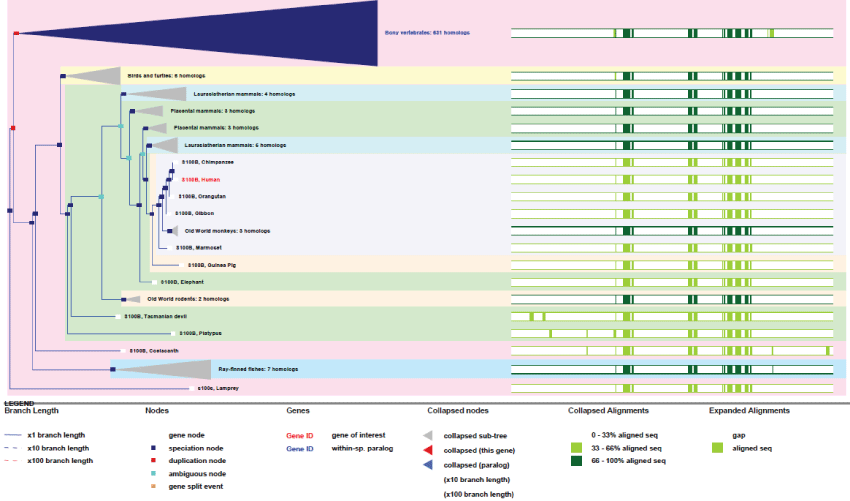 Figure 13: Phylogenic tree of S100B.View Figure 13
Figure 13: Phylogenic tree of S100B.View Figure 13
Also evolved as a family of tissue-specific expressed genes, the S100B gene represents a distinctive example from the others studied here because it has been duplicated many times to create variants rather than using a differential splicing strategy.
In higher eukaryotes, mitochondria have evolved very far from the original and basic concept of a simple factory, producing ATP for the cell. As we have seen, mitochondria has developed a wide variety of essential functions to ensure cell homeostasis, growth and differentiation.
We did not focus here on the thermogenesis, and the UCP proteins, which represents also and by itself a great and crucial improvement in biology of cell along evolution. We will neither insist about all the numerous links existing between mitochondria and pathologies which concern cancers, neuropathies (ataxia, ophthalmia, deafness), encephalopathies, myoclonic epilepsy and many others diseases [51]. However, when considering all the known mitochondrial functions, it is easy to conceive how many diseases can originate from mitochondria dysfunctions.
The most astounding point when considering mitochondrial functions is to discover how complex and tightly coupled connections have to exist between mitochondria and the cell, generally at the nuclear but specially at the transcription levels. Neither is it surprising to discover today how some proteins can switch from one compartment to another, depending on pre- or post-translational modifications.
However, at the genetic and evolution levels, we can observe that there was apparently no need for creating and amplifying several of the genes, as many stayed unique. Most genes directly implied in mitochondrial functions are found in yeast and bacteria, but because they evolved and contained thereafter many exons, it resulted in the production of many variants from unique genes.
Whatever, it appears that the mitochondrial genome remains the oldest trace of the mitochondrial functions and evolution. It may represent the last traces of the gene transfer to the nucleus, with therefore the improvement in eukaryotic encoding system (exons) resulting in optimization of all mitochondrial processes through better "addressing" of the energy.
The last sign of evolution is the regulation of energy usage, with short, mid and long term regulations and adaptations such as biological tendency for economy, cell size and adaptability which appeared last (Largen, S100...).
One might anticipate that mtDNA could one day disappear from mitochondria when all these metabolic machineries would be provided by the nucleus. The reason to refute this possibility is the existence of the link between OXPHOS system physiology, mtDNA and contact sites between ER and mitochondria. This is essential to keep that way an appropriate co-regulation system between cell and mitochondria.
Among the particularities of mitochondrial genetic evolution, improvement of energetic brain function resulted in increased capacities of neurons to analyse, integrate and communicate with themselves, organs and individuals. This is probably the last main great improvement of primate evolution.
The conclusion could be that "to become larger and bigger energy-consumer, it was is not necessary to increase the number of genes for respiration. It is apparently much better to improved signal transduction, regulation and adaptation of mitochondrial functions and mass, with a small set of specialized genes and proteins". However, understanding how the initial mitochondrial gene/DNA succeeded to reach the nuclear compartment remains an amazingly interesting opened question.
I am deeply thankful to Dr. Jean Gagnon for helps in writing the manuscript. I am also extremely grateful to Dr. Yuliya Lyubenova Dimitrova for consistant helps in work.