NF-κB essential modulator (NEMO) syndrome is an immunodeficiency disease. NF-κB proteins, which regulate the expression of genes that moderate important physiological processes, are called regulatory of cell homeostasis. NEMO is a protein in the IKK inhibitor complex that many organ systems normally do not grow. Cells (as well as organ and tissues) do not grow proteins they express proteins. The disease occurs due to mutation in the IKBKG gene. The IKBKG gene, located in the Xq28 chromosomal region or located in the X chromosome. The disease indicates an impairment of NF-κB activation and the initial treatment of NEMO is very difficult. About 70-80% of patients have similar DNA rearrangements. Epilepsy is observed in about 50% of patients with these disorders. Therefore, there is little information about the NEMO disease and more research is needed to further examine the syndrome.
NF-κB, NEMO, Immunodeficiency, IKK, IKBKG
NF-κB proteins, which regulate the expression of genes that moderate important physiological processes (immunity, inflammation, death, and cell proliferation), are called regulatory of cell homeostasis. NF-κB is regulated by a number of different stimuli [1]. The NF-κB factors include five proteins: RelA, RelB, c-Rel, p100, and p105. All of these have relative homology (RHD), which is involved in binding to DNA and interacting with lκB inhibitors [2,3]. Three groups of Rel homology domain A, B, and C have the transcription activation domain, but other groups (p100 and p105) do not have transcription activation domain (TAD). In fact, these proteins are a precursor to NFκB and the p50/RelA subunit is expressed in most cells [4]. Therefore, the functions of this transcription factor (NF-κB) are important to the central nervous system (CNS). This transcription factor plays a regulatory role in the response to synaptic transmission and neuronal survival. In general, diseases caused by NF-κB signaling mechanisms lead to many abnormalities in the immune system, vascular system, skin, bones, and CNS [5,6]. One of the most important features of NF-κB transcription factors is their association with IKB inhibitors [1]. This protein inhibitor is found in the cytoplasm cell and consists of three members: IκBα, IκBβ, and IκBε [1]. In the N-terminal of these inhibitors, the protected sequence contains two Serine residues, which can be altered by phosphorylation [7]. Serine phosphorylation is performed by the IκB kinase complex (IKK). The main components of this kinase complex include IKK1 and IKK2 subunits and regular subunit, NEMO [8]. NEMO is a binding domain (NBD) containing the IKK interaction sequence in the N-terminal, NEMO ubiquitin binding in the mid-section, and the Z-finger (ZF) structure in the C-terminal [9].
IKBKG mutations and NEMO defect were first detected in 1999. This syndrome is a genetic mutation associated with the defect of the NEMO protein which many organ systems normally do not grow including immune deficiency. The NEMO syndrome is a disease caused by genetic mutations in the X-linked gene. The syndrome is observed in different people in different ways. The most common symptoms are skin disease and bacterial infections [1]. NEMO is an IKK regulatory subunit that has affinity to polyubiquitinated chains and leads to the use of upstream activators and catalytic IKK activity. Cells that do not activate NEMO NF-κB are eliminated through the main pathway [10].
Bloch-Siemens syndrome is a genetic disease associated with chromosome X that occurs in males at an early stage of development and is fatal before the second trimester. But in females, due to the inactivation of chromosome X, the reduction of genetic defects and pathology is more severe and variable [1,11,12].
One of the most common features of this syndrome is the presence of severe dermatosis, which often begins two weeks after birth and lasts for months and years. In this disease, the first symptoms are whiteness of skin and inflammatory response. Afterward, hyperkeratoscopic lesions are created (the melanin uptake eliminates parts of the hyperpigmentation). Then, there are the lines of Blaschko, which often disappear in the second decade, but it is possible to observe skin allergies and lack of hair follicle in adults. The patients can suffer from dental, ocular, skin, and nervous problems, as well as nerve dystrophy in very rare cases. More than 80% of patients are observed to have inappropriate, incomplete, or excess teeth [13]. Approximately 35% of patients have eye problems mostly abnormalities of the retina; in certain cases, acute blindness may occur [14]. Neurological abnormalities are found in about 30% of these patients. Problems like epilepsy, skin disease, hemiparesis, cerebellar ataxia, microsphere, mental retardation, and spasticity in early life are associated with rare cases of CNS disorders and can be fatal. Seizures caused by severe cerebrovascular injury cause thalamus bleeding, ischemia, and necrosis in both hemispheres [15].
The Bloch-Siemens encoding gene is located in the chromosomal region Xq28 [16]. NEMO is in a position where its gene is mutated. PCR analysis of the NEMO gene in male IP (Incontinentia Pigmenti)-embryonic cells reveals a lack of proliferative fragments of exon 10-4 [17]. About 70-80% of patients suffer from loss of exon NEMOdel. This process results from nonallelic homologous recombination (NAHR) between two sequences of MER67B at the upstream of exon 4 and downstream of exon 10 NEMO (Figure 1) [1].
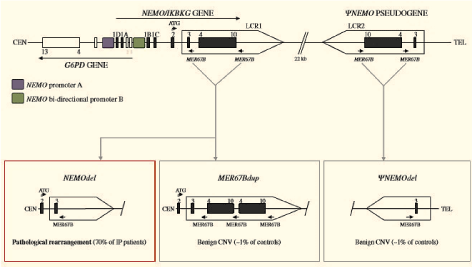 Figure 1: DNA rearrangements of the NEMO and ψ-NEMO loci. In the upper part of the figure, the genomic organization of the NEMO gene and the pseudogene ψ-NEMO (IP locus) is shown together with the genomic organization of the neighboring gene G6PD, which shares promoter elements with NEMO. In the lower part of the figure are shown the main rearrangements occurring in the IP locus. Among them is the recurrent one that deletes exon 4-10 of NEMO in approximately 70% of IP patients (Red frame) [1]. View Figure 1
Figure 1: DNA rearrangements of the NEMO and ψ-NEMO loci. In the upper part of the figure, the genomic organization of the NEMO gene and the pseudogene ψ-NEMO (IP locus) is shown together with the genomic organization of the neighboring gene G6PD, which shares promoter elements with NEMO. In the lower part of the figure are shown the main rearrangements occurring in the IP locus. Among them is the recurrent one that deletes exon 4-10 of NEMO in approximately 70% of IP patients (Red frame) [1]. View Figure 1
The NEMO map or IP location in the centromeric region overlaps with the glucose-6-phosphate dehydrogenase (G6PD) gene. The promoter region protected in these two genes is Promoter B. But the special promoter for NEMO is Promoter A [18]. In the telomeric region, NEMO is in the part of 35/7 kb, which has a low or non-functional copy called pseudoNEMO (ψ-NEMO). The two LCRs-functional gene (LCR1) and false gene expression (LCR2) are homologous and susceptible to recombination. Evidence suggests that the IP gene site undergoes NAHR production, pathological rearrangement (NEMOdel), or MER67Bdup and pseudoNEMOdel (Figure 2) [1,18].
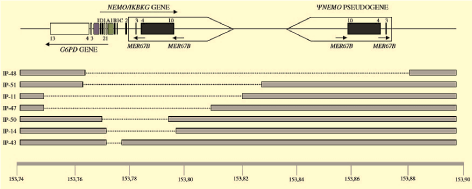 Figure 2: Chromosomal deletions affecting the NEMO gene in IP. The various deletions that have been identified are compiled. In several instances, G6PD is also affected. The largest one identified so far (deletion IP-48) also eliminates ψ-NEMO [1,18]. View Figure 2
Figure 2: Chromosomal deletions affecting the NEMO gene in IP. The various deletions that have been identified are compiled. In several instances, G6PD is also affected. The largest one identified so far (deletion IP-48) also eliminates ψ-NEMO [1,18]. View Figure 2
Also, some other findings indicate that pseudoNEMOdel and MER67Bdup variants are known as risk alleles for the IP [1]. Findings by Ardia, et al. show the sequencing exchange between LCR1 and LCR2, which refers to the occurrence of inverse events from the opposite direction. Additionally, evidence suggests that recombination by NAHR takes place between the two LCRs. Polymorphic changes in pseudoNEMOdel or its point mutation causes pathogenic IP mutations [19-21]. The rearrangements include the removal of NEMO and ψ-NEMO and G6PD. In some cases, NHEJ (non-homologous end-joining) is shown to produce benign and pathologic alleles, NHEJ, and the Alu-Alu recombination. NEMO reinsertion in IP leads to short versions of NEMO [19,21,22].
There is an exception namely microdeletion (deletion of the amino acid K90). The mutation in K90 is associated with intense IP status, which is not necessary for the interaction of NEMO with catalytic subunits of the IKK. However, the deletion of aa in this section is sufficient to disrupt the amino acids E89, F92, and L93 and the remaining D738, T735, and F734 (IKK2). It is worth noting that there are a few missense mutations in the NEMO of these patients (Figure 3) [23,24]. The mutation in the A323P causes the development of IP and CNS disorders. There is evidence that these mutations affect the signaling pathways of NF-κB. The mutation in E57K stimulates the interaction of NEMO and TRAF6, creating a dual interaction between NEMO and TRAF6, which does not interact with IKK [1,23,24].
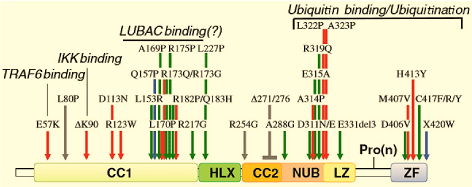 Figure 3: NEMO missense mutations and short internal deletions of NEMO found in pathology. Red arrows indicate IP-related missense mutations, green arrows indicate EDA-ID-related missense mutations, blue arrows indicate OL-EDA-ID-related missense mutations, and gray arrows indicate ID-related missense mutation. A short internal deletion of NEMO associated with ID is also indicated with a gray bar. Compiled from Refs. [19,37,45]. Formally and putatively (question mark) demonstrated molecular defects are also indicated. View Figure 3
Figure 3: NEMO missense mutations and short internal deletions of NEMO found in pathology. Red arrows indicate IP-related missense mutations, green arrows indicate EDA-ID-related missense mutations, blue arrows indicate OL-EDA-ID-related missense mutations, and gray arrows indicate ID-related missense mutation. A short internal deletion of NEMO associated with ID is also indicated with a gray bar. Compiled from Refs. [19,37,45]. Formally and putatively (question mark) demonstrated molecular defects are also indicated. View Figure 3
Although there are no phenotypic symptoms in these patients, the initial treatment of NEMO is very difficult. As stated above, 70-80% of patients have similar DNA rearrangements.
The statistics show that it only affects 50% of the patients. Epilepsy is observed in about 50% of patients with these disorders. Reports show that appropriate treatments are sometimes ineffective. It should be noted that skin anomalies in the disease of the IP are similar to psoriasis. However, it is not yet clear whether inhibiting a process can lead to the elimination of mutated cells [25-27].
According to findings on mutated NEMO, men with immunodeficiency syndrome are not associated with ectodermal dysplasia (EDA) (Figure 3). These patients do not show a specific infection; therefore, they can be differentiated from EDA-ID patients. At the moment, it is difficult to understand that NEMO mutations do not cause EDA. Mutations that contribute to protein expression have a great impact on cellular immunity. The NEMO frameshift mutation in alanine 37 amino acids is located near methionine 38 and causes immunodeficiency without any symptoms of EDA. However, this problem is interesting because it involves two mutations occurring only in an immunodeficiency, which results in short deletions in 276-271 and 373-353 domain, which have not been determined yet [28-33].
As stated, Bloch-Siemens syndrome is due to the lack of NEMO mutation. NEMO hypomorphic mutations cannot completely inhibit NF-κB activation. Evidence suggests that the X-linked pathology is associated with EDA-ID. It affects the male, but in rare cases, it also it affects females that is mildly similar to IP. The characteristics of this disease (EDA-ID) are severe immunodeficiency and skin problems, including problems in teeth, sweat glands, and hair [34,35].
The patients suffer from frequent bacterial and viral infections. The most common pathogens are S. pneumoniae and S. aureus. In most cases, chronic mycobacterial infections are observed, and some of these patients also suffer from HSV encephalitis [1]. The disease is associated with poor prognosis and progress. It stimulates adaptive immunity in some patient; defects in the production of specific antibodies have been observed in hyper IgM syndrome [36,37].
Defective cell B causes CD40 disorder. In rare cases, T-cell deficiency has been reported, which affects mycobacterial infections as well as viruses. However, its molecular and clinical diagnosis is difficult [38]. Colon inflammation has been reported in some EDA-ID patients. The disease is similar to Crohn's disease, but it is still unclear whether it is due to mycobacteria or whether EDA-ID causes a defect in the Nod2 signaling pathway. Also, this EDA-ID leads to problems such as pale hair, lack of eyebrows and eyelashes, severe oligodontia, and the absence or reduction of sweat [1,39-42]. Studies using biochemical analysis show that these abnormalities lead to a mutation in the signaling pathway of ectodysplasin EDA receptor, which is related to EDA. The receptor of edema ectodysplasin is bound to IKK/NF-κB by NEMO. The OL-EDA syndrome is one of the rare cases that causes a defect in the RANK signaling pathway. In fact, this syndrome is caused by disturbances in the signaling pathway of VEGFR3 [43,44].
NEMO mutations in EDA-ID are more dangerous than IP mutations, and survival rate is lower in males. Hypomorphic mutations show a change in NEMO activity, which indicates heterogeneity of the EDA-ID phenotype. The frameshift and nonsense mutations cause EDA-ID. The mutations in some cases result in the removal of NEMO ZF. In most patients, deletions are seen through missense mutations [37,45,46].
Moreover, the factor that affects the oligomerization and activation of NF-κB is due to a mutation in the EDA-ID or an A288G mutation in the CC2 region of the NEMO [47]. On the other hand, the mutation associated with OL-EDA-ID or X420W causes severe protein deficiency [47,48]. However, it is still unclear whether the NEMO expression defect is responsible for the OL-EDA-ID. It is also noteworthy that OL-EDA-ID is due to the Q157P mutation in the NEMO region, leading to EDA-ID or IP.
If the patients become infected, the plasma protein C concentration does not increase (due to a defect in the TLR-signaling pathways); therefore, they should be treated with intravenous antibiotics. The infection in EDA-ID patients can be dangerous; therefore, the recommended method is to restore some protective functions of the allogeneic HSCT. But this method of treatment also has problems and is not always successful. In patients who show bowel inflammation before the transplant, a transplant may worsen the condition [47,49-51].
Diseases of the NEMO mutation indicates an impairment of NF-κB activation. Recent findings indicate that NEMO plays a specific protective role in blood vessels and may lead to IP-related abnormalities in the event of its functional impairment. This provides molecular targets for new therapies. There is little information about the NEMO disease and more research is needed to further examine the syndrome.
No funding was received for this review.
Mohammad Reza Zinatizadeh conceived the project, wrote the outline and first draft, integrated the appraisal, and finalized and submitted the manuscript. Zahra Masoumalinejad helped in the initiation, outline and direction, appraisal, and final approval of the manuscript. Azim Nejatizadeh contributed to the critical appraisal and final approval of the manuscript. Mohammad Shekari helped in the initiation, outline and direction, appraisal, and final approval of the manuscript. Farzaneh Parnak and Faeghe Zaree contributed to the initiation, outline and direction, appraisal, and final approval of the manuscript. All authors read and approved the final manuscript.
The authors declare that they have no competing interests.