Splicing is a part of mRNA maturation process when exons of pre-mRNA transcript are joined in multiple ways. In case of alternative splicing, some exons may be excluded from the final mRNA, and the resulting ensemble of mRNAs creates different protein isoforms, allowing multiple proteins to be coded by a single gene. To detect new events of alternative splicing in human blood cells, we used RNA-seq technology. A pipeline based on Python "Scikit-Learn" library was developed for efficient splicing events calling. We found 8728 potential candidates, 20 of which were selected for RT-PCR and 8 were finally confirmed as novel events in 7 genes (MPPE1, CTDP1, ENOSF1, SEH1L, TXNL4A, C18orf1, and ME2). SVMOneClAS are freely available from the https://github.com/alsokolov-dev/SVMOneClAS
Alternative splicing, Machine learning, Support vector machine, RNA-seq
Alternative splicing occurs generally in eukaryotes and is considered to be a key factor in protein functional complexity and diversity [1]. It has been observed that ~95% of the multiexon genes in humans are subject to alternative splicing events [2]. Among different modes of alternative splicing, exon skipping appears to be the most common mode characterized by exon inclusion or exclusion depending on mRNA production conditions [1]. At the same time alternative 5' or 3' splice sites and intron retention modes are demonstrated in cancer cells [3]. Several protein products can result from one gene by the use of alternative mRNA splicing, and some of these splice isoforms can lose or gain a function. Therefore, apart from the protein diversity, alternative splicing is a cause of abnormal splicing variants resulting in functional consequences. This way, aberrant mRNA transcripts are implicated in a large number of human genetic disorders. Furthermore, hundreds of genes are found to be abnormally spliced in cancer cells, which is functionally important for cancer development [4-6].
Recent discoveries and technological advances have given an opportunity to deeply investigate alternative splicing events providing new insight into physiological and biochemical network in different systems. Such studies not only reveal mechanisms of cellular processes regulation but also clear the way to the development of new therapeutic strategies to disorders treatment. Pathogenesis of a number of neurodegenerative, cancer, blood, inflammatory, and hemolytic disorders was found to be associated with alternative splicing [7,8]. For example, study of stress responses mechanisms in brain and blood described stress-induced changes associated with alternative splicing patterns of acetylcholine pre-mRNA and functional acetylcholine properties [9]. Another research in the field of thrombosis and hemostasis discovered genes producing alternatively spliced protein isoforms that can break some coagulation cascades and affect blood clotting [10].
Current studies of alternative splicing and its mechanisms showed an important role of such events in the cellular processes; however, characterization of new alternative splicing events as well as their tissue specificity is still in high demand. Here a combination of PCR, Sanger sequencing, and RNA-seq data analysis was successfully applied to find new events of alternative splicing.
In order to identify new events of alternative splicing, we used a «One Class Support Vector Machine» algorithm [11-15]. This algorithm is often used for novelty detection, when one is able to decide whether a new observation (in our case a new alternative splicing event) belongs to the same distribution compared to existing observations (canonical events of alternative splicing).
The pipeline workflow was as follows: A data set of n canonical splicing events described by p features was considered; new observations (new events of alternative splicing) to that data set were added. Then the question was: do these events come from the same distribution or more precisely are these events so similar to the canonical that we cannot distinguish it? This self-taught algorithm identified a rough, close frontier, delimiting the contour of the canonical splicing events distribution, plotted in embedding p-dimensional space. Then, if new events of alternative splicing lay within this delimited subspace, they are considered as potential new events of alternative splicing. On the other hand, if they lay outside, we can say that they are abnormal with a given confidence in our assessment.
The pipeline outline is shown on Figure 1A. 4 types of kernels were used to best fit the data («linear», «poly», «rbf» and «sigmoid»). Data set was divided on train and test sets for purposes of cross-validation procedure. The training set is used to train the classifier with two parameters: Gamma (kernel coefficient for "rbf" and "poly" types of kernels functions) and degree (degree of kernel function). The test set is used to evaluate the accuracy of each classifier and then kernel function and parameters gamma and degree that gave smallest value of error on cross validation step was used for subsequent analysis.
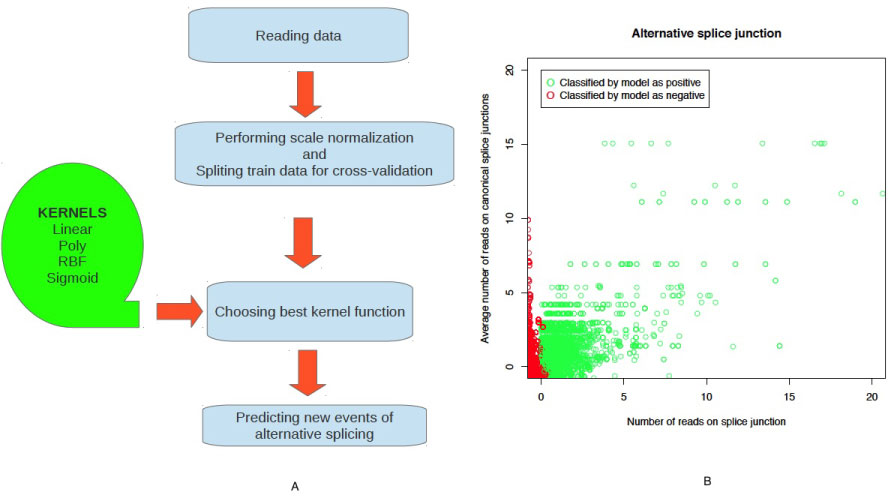 Figure 1: A) Pipeline outline; B) New events of alternative splicing. View Figure 1
Figure 1: A) Pipeline outline; B) New events of alternative splicing. View Figure 1
Whole blood RNA isolated from an individual N, whose genome was sequenced previously [16,17], was used to prepare a library according Illumina protocols. Illumina GAIIx RNA-seq reads generated were mapped to reference human genome and splice junctions (Table S1, Table S2, Figure 2 and Figure S1) (we used RefSeq database of known exons to construct all possible exon junctions) of human genome using Bowtie2 [18,19].
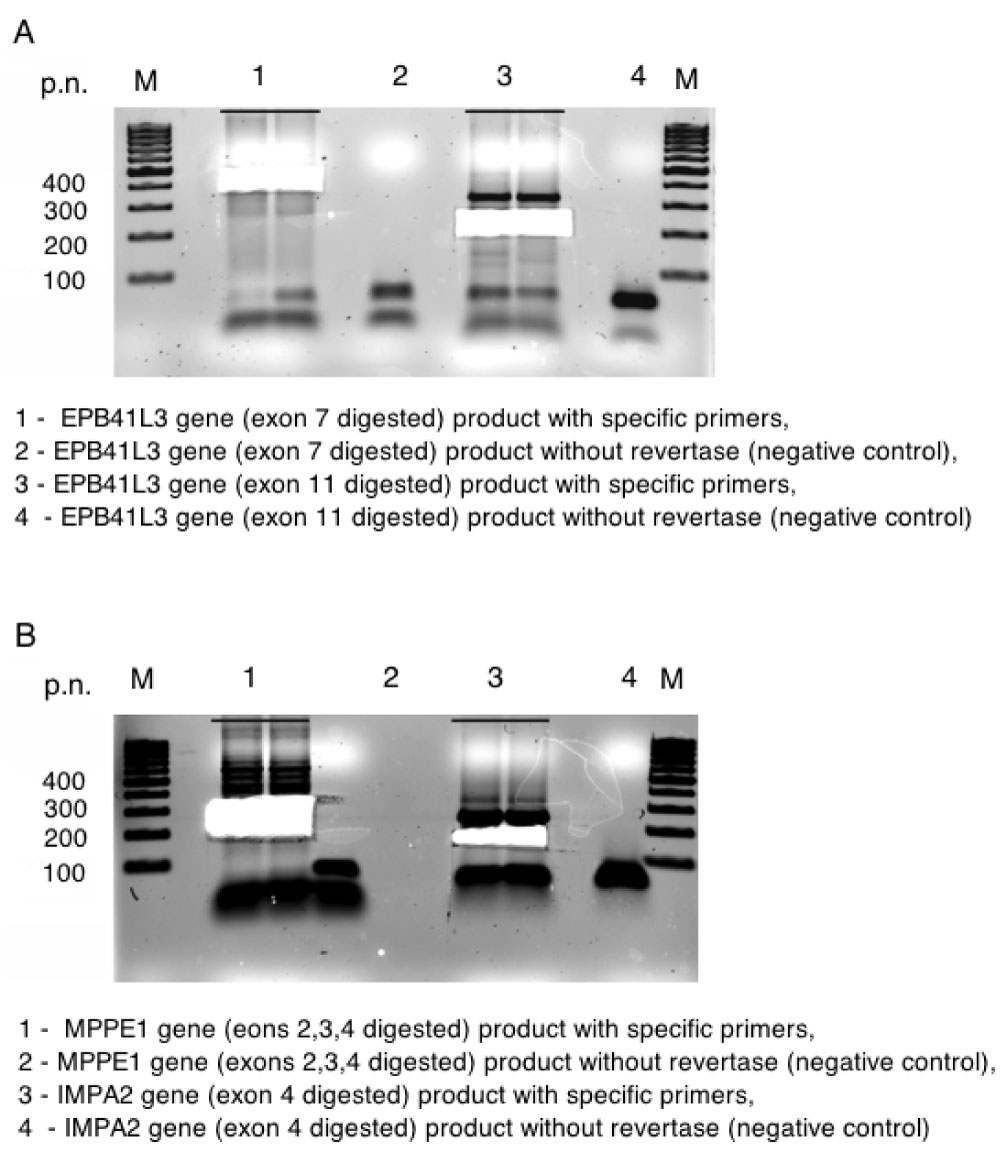 Figure 2: Finding and confirming new events of alternative splicing.
Figure 2: Finding and confirming new events of alternative splicing.
A, B) Electrophoresis of PCR products for genes EPB41L3 and MPPE1, IMPA2 resp. View Figure 2
 Figure S1: Results of mapping reads on genome. View Figure S1
Figure S1: Results of mapping reads on genome. View Figure S1
Table S1: Reads mapped on genome. View Table S1
Table S2: Reads mapped on splice junctions. View Table S2
We used 4 parameters for each splicing event: number of reads at this splice junction, average number of reads at all canonical splice junctions of this transcript, gene length, and a number of exons. Compared with papers that use Support Vector Machines (SVM) to find new events of alternative splicing [20-23], we didn't use sequence information, which makes our algorithm more appropriate for searching for different alternative splicing events between various tissues or conditions of the same organism. We found 8728 (28% of all possible events) potential candidates (Figure 1B), 20 of which were selected for subsequent analysis Table S3 (none of those events were found by cufflinks).
Table S3: None of those events were found by cufflinks. View Table S3
To confirm new events of alternative splicing we performed RT-PCR with specific primers, PCR products were fractionated in agarose gel, amplicons of expected size were eluted and sequenced (Sanger).
Only 15 events out of 20 tested produced fragments of expected size and 11 were proved by sequencing to be alternative splicing (Figure S2, Figure S3, Figure S4, Figure S5, Figure S6, and Figure S7). Next, we translated DNA sequences of the amplicons and ran corresponding ORF against UniProt database. This procedure filtered out additional 3 events. The rest 8 events (referred as "8 events") were localized in 7 genes, i.e. MPPE1, CTDP1, ENOSF1, SEH1L, TXNL4A, C18orf1, ME2 and most of them are already described in HAVANA database. Then we isolated RNA from whole blood of 10 healthy individuals of Slavic origin and performed RT-PCR with specific for "8 events" primer sets. Among 10 tested patients, 8 carried full set of "8 events" and the rest 2 shared 6 out of "8 events" (Figure S7 and Figure S8).
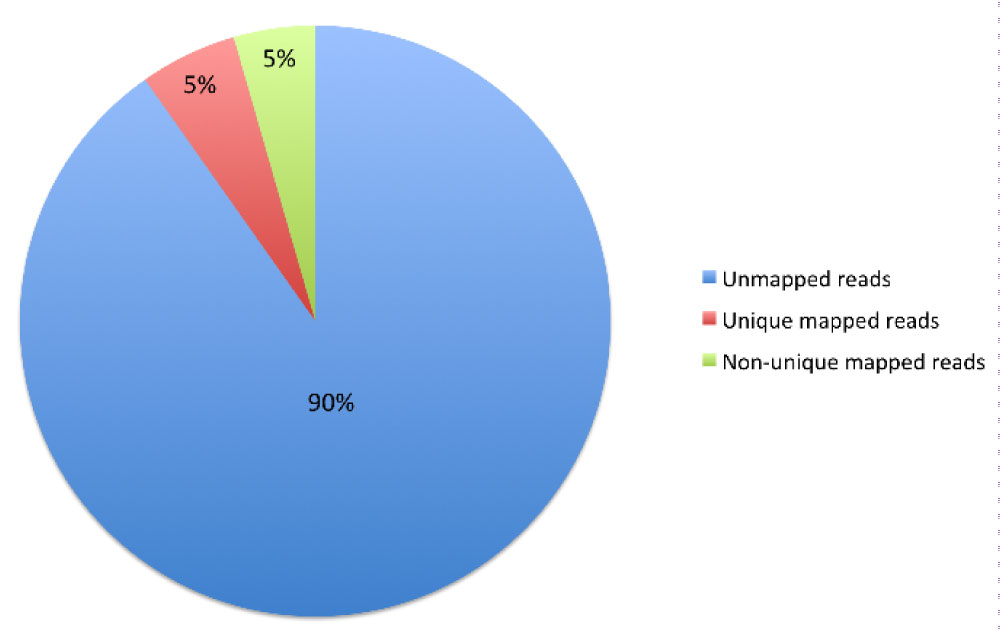 Figure S2: Results of mapping reads on splice junction. View Figure S2
Figure S2: Results of mapping reads on splice junction. View Figure S2
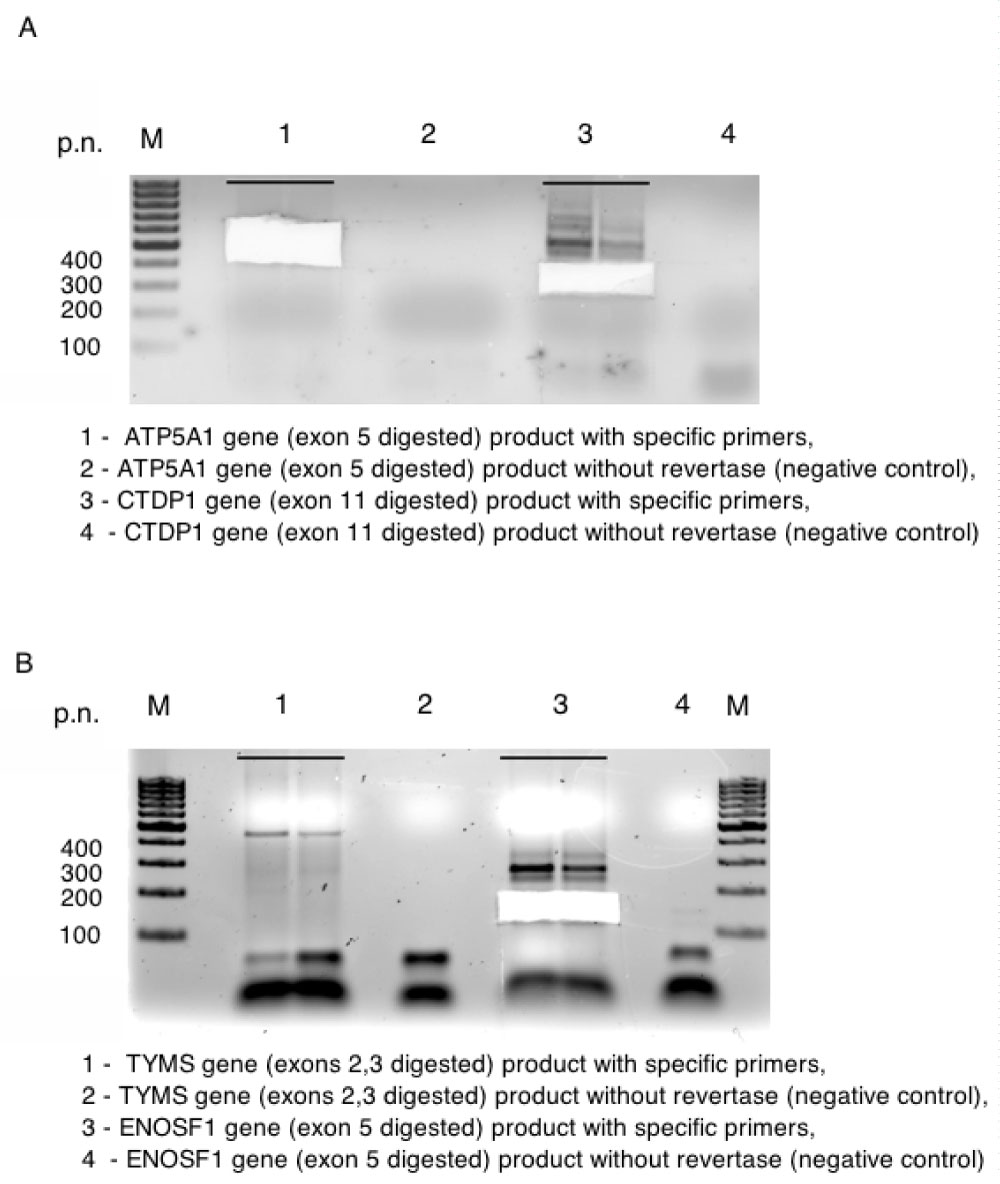 Figure S3: A, B Electrophoresis of PCR products for genes ATP5A1, CTDP, TYMS and ENOSF1 resp. View Figure S3
Figure S3: A, B Electrophoresis of PCR products for genes ATP5A1, CTDP, TYMS and ENOSF1 resp. View Figure S3
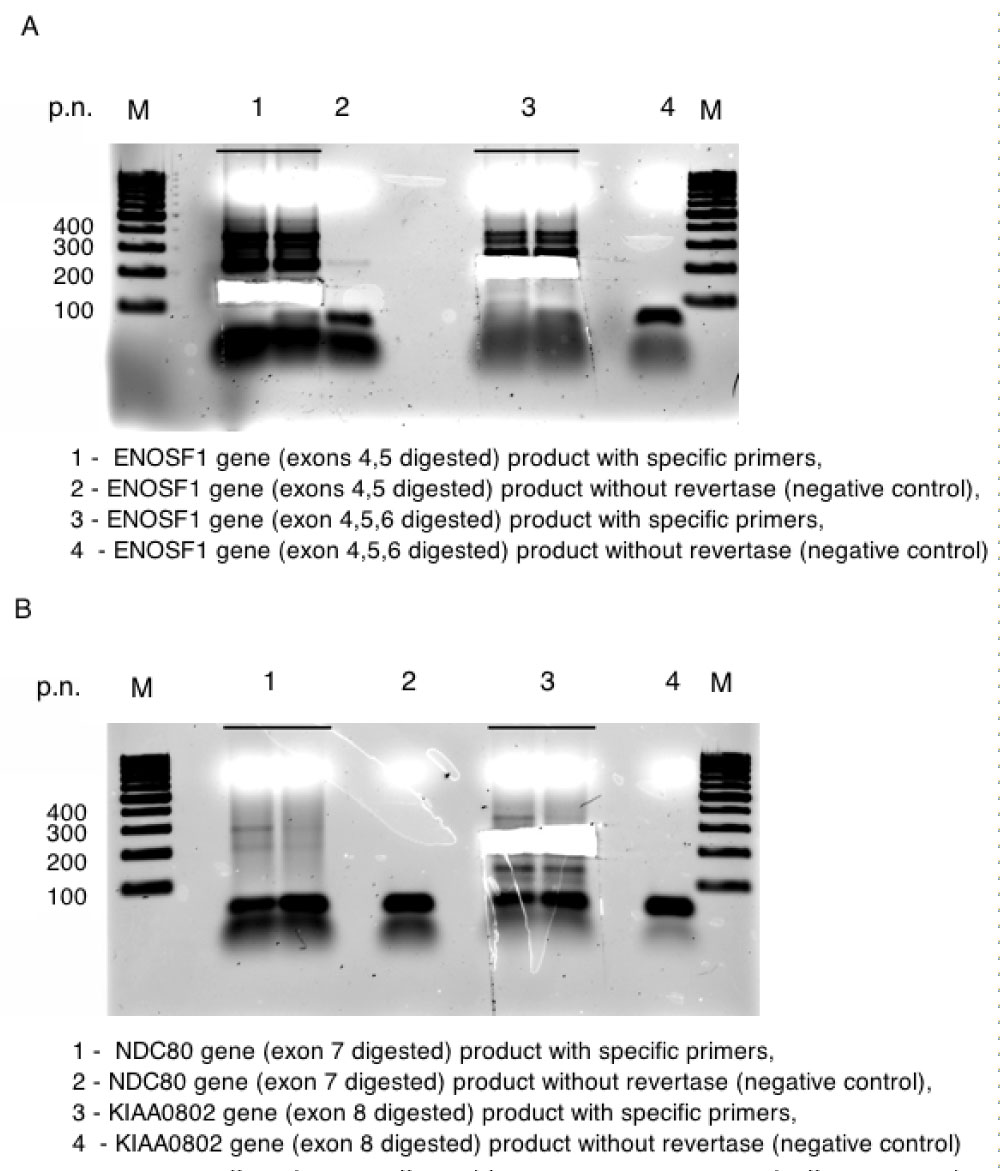 Figure S4: A, B Electrophoresis of PCR products for genes ENOSF1, NDC80, and KIAA0802 resp. View Figure S4
Figure S4: A, B Electrophoresis of PCR products for genes ENOSF1, NDC80, and KIAA0802 resp. View Figure S4
 Figure S5: A, B Electrophoresis of PCR products for genes SEH1L, ATP5A1, TXNL4A resp. View Figure S5
Figure S5: A, B Electrophoresis of PCR products for genes SEH1L, ATP5A1, TXNL4A resp. View Figure S5
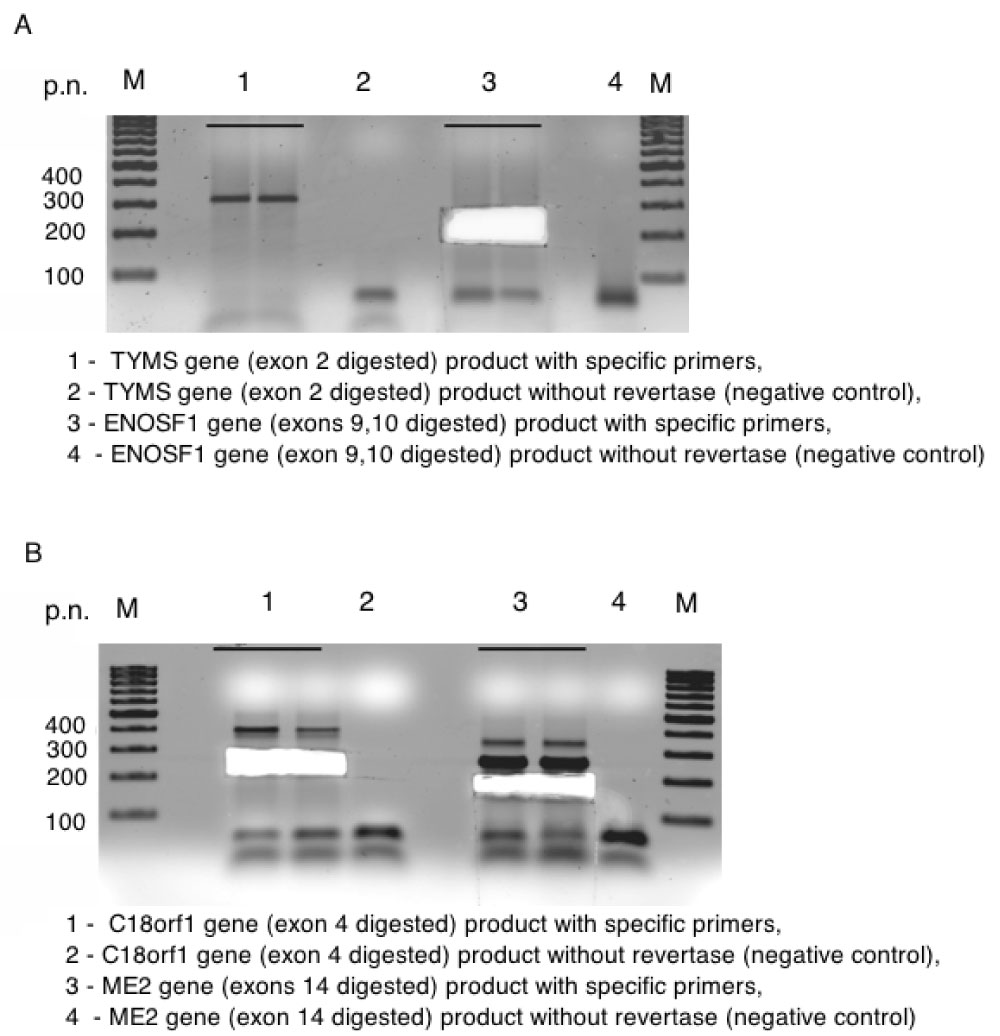 Figure S6: A, B Electrophoresis of PCR products for genes TYMS, ENOSF1, C18orf1 and ME2 resp. View Figure S6
Figure S6: A, B Electrophoresis of PCR products for genes TYMS, ENOSF1, C18orf1 and ME2 resp. View Figure S6
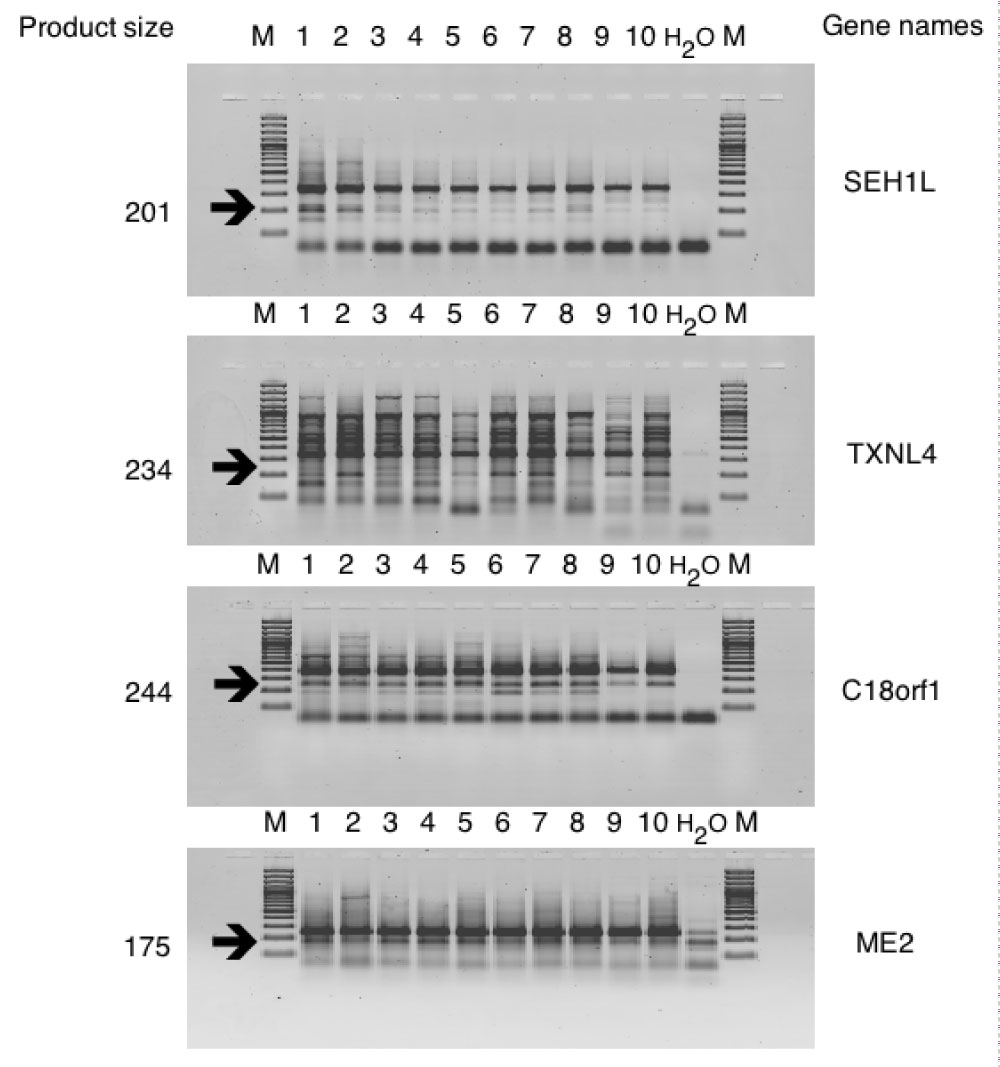 Figure S7: Confirming of new splice events by RT-PCR with specific primers on 10 healthy blood donors 1,2,3,4,5,6,7,8,9,10 - samples numbers; M - Marker; H2O - Negative controls; arrows indicate specific product size. View Figure S7
Figure S7: Confirming of new splice events by RT-PCR with specific primers on 10 healthy blood donors 1,2,3,4,5,6,7,8,9,10 - samples numbers; M - Marker; H2O - Negative controls; arrows indicate specific product size. View Figure S7
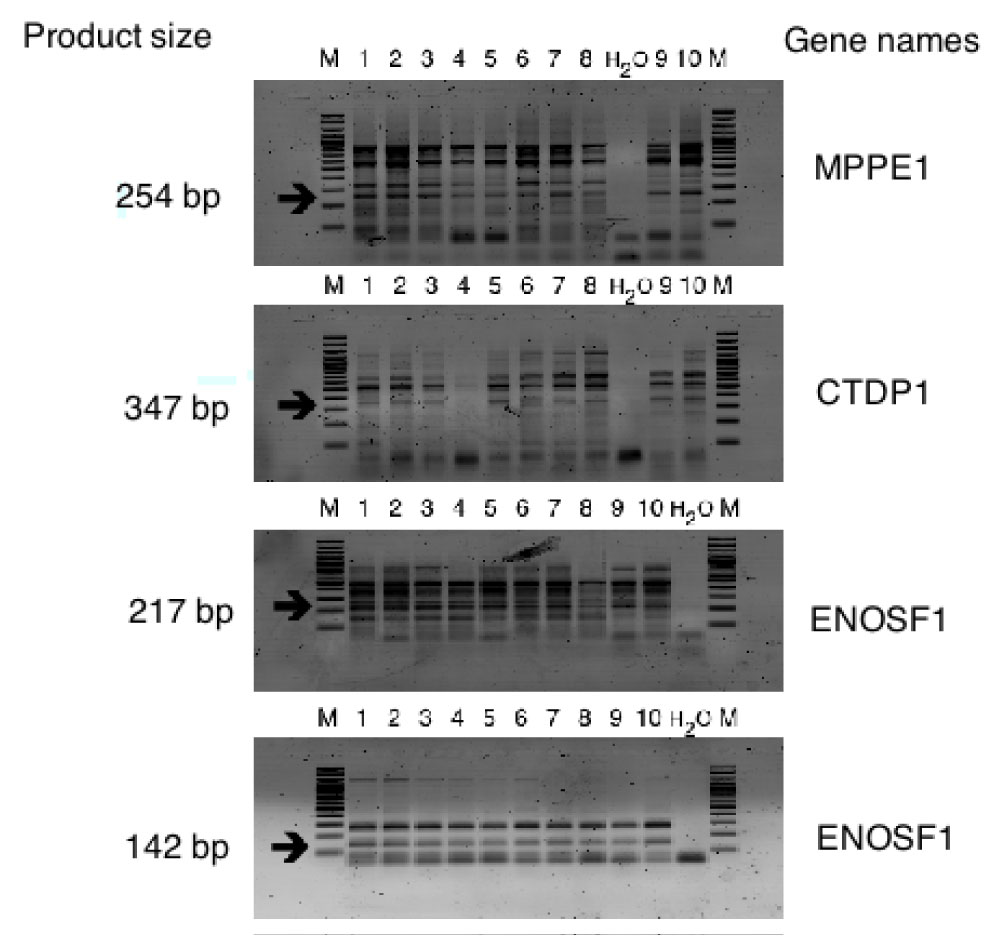 Figure S8: Confirming of new splice events by RT-PCR with specific primers on 10 healthy blood donors 1,2,3,4,5,6,7,8,9,10 - samples numbers; M - Marker; H2O - Negative controls; arrows indicate specific product size. View Figure S8
Figure S8: Confirming of new splice events by RT-PCR with specific primers on 10 healthy blood donors 1,2,3,4,5,6,7,8,9,10 - samples numbers; M - Marker; H2O - Negative controls; arrows indicate specific product size. View Figure S8
All new events represented an exon skipping mode of alternative splicing. When exon skipped, protein may miss some specific sites or domains after translation. In our case proteins may miss metal-binding sites (as for genes MPPE1, ENOSF1, ME2), active sites (as for genes ENOSF1, ME2), topological domain (as for gene C18orf1), or some particular sequences, for example, repeat 55-96 (as for SEH1L gene).
The authors thank K.G. Skryabin, P. V. Mazin and M.V. Kovalchuk for the support and careful attention to the project. This study was supported by the Ministry of Education and Science of Russia (project № 14.740.11.0759) and by the Scientific Research Agreement №248 with the Institute of Biomedical Chemistry RAMS (a part of the Human Proteome Project, Russia).