Autism Spectrum Disorder (ASD) comprises a complex of neurodevelopmental disorders primarily characterized by deficits in verbal communication, impaired social interaction and repetitive behaviors. The complex genetic architecture of ASD encompasses profound clinical heterogeneity, which poses huge challenges in understanding its pathophysiology. We conducted a large scale association analysis of the MSSNG whole genome sequencing data to elucidate potential modifiers of ASD severity. Using linear regression, we have directly tested the association between 6,198,166 SNPs and Vineland Adaptive Behavior Scale Scores a standardized metric for measuring severity across multiple ASD spectra.
The most significant variants direct us to a significant haplostretch chr3p21 (pval 3.68e-12) of SNPs, n = 132) containing variants on chromosome 3 including a highly interesting nonsynonymous SNV rs11539148 within the QARS gene (NM_001272073:c.A821G:p.N274S MAF = 0.0391) a glutaminyl-tRNA synthetase coding gene crucial in brain development. Furthermore, we analyzed eQTLs for QARS, and found decreased expression across several datasets, a result consistent with the observed effect. The effect further potentially explains differences in significant changes in head circumference. To leverage the size of the region we conducted a pathway enrichment analysis of the set of highly significant loci. The most significant categories include brain development and structural component of the myelin sheath. Genes categorized as neurological, developmental and immune-related constitute 65% of all the genes contributing to these pathways.
Our analysis has detected a region that may be a hallmark of severity in ASD. As the genetic predisposition may be different for almost every ASD individual, understanding the common mechanisms for endophenotypes may help elucidate ASD causal mechanisms.
Our analysis has detected a region that may be a hallmark of severity in ASD. As the genetic predisposition may be different for almost every ASD individual, understanding the common mechanisms for endophenotypes may help elucidate ASD causal mechanisms.
Autism spectrum disorder (ASD) affects 1 in 63 of all children born in Europe, North America and other developed regions, and is defined by persistent alterations in social communication and interaction alongside restricted, repetitive patterns of behavior, interests or activities [1,2]. It causes significant impairment in social, occupational and other important areas of functioning. Psychiatric comorbidity is common, including attention deficit hyperactivity disorder (ADHD), affecting up to 30% of children with ASD. Heritable factors account for at up to 80% of ASD risk with the remainder attributable to environmental factors acting alone or through interaction with genetics [3].
Tremendous progress has been made in understanding the genetic underpinnings of Autism Spectrum Disorders (ASD) with potential variants usually covering the entire spectrum of mutations from single nucleotide variants to loss/gain of copy number effects. In addition to inherited variants, genomes of probands are enriched in de novo genetic variants [3,4]. Such variants are estimated to be causal in 10-30% of cases with ASD. The estimated contribution of common variants taken cumulatively, however, ranges from 15% to 50% [5]. Moreover, estimates of both penetrance and expressivity of well-established risk variants vary widely. The genetic architecture of ASD clearly consists of a wide spectrum of risk alleles. At one extreme are the dominant acting de novo variants that carry high risk and are rarely carried by asymptomatic parents [6]. At the opposite extreme are many common 'polygenes' that individually exert subtle influences on risk. A major role of genetic factors in the causation of ASD has been supported by genetic epidemiologic studies, molecular genetic studies and the co-occurrence of autistic syndromes in monogenic conditions. Nevertheless, we still cannot pinpoint the precise gene variants that would explain the incidence; hence, the predictive power of genetic for ASD is small. A multifactorial etiological model for ASD is being increasingly recognized. Consequently, numerous efforts to identify genes associated with ASD risk have been undertaken with the aim of inferring molecular pathways or surrogate markers associated with clinical manifestations of ASD [6]. While initial estimates suggested between 350 and 400 autism susceptibility genes, more recent statistical models predict that well over 1,000 genes may eventually be associated with ASD (Simons Foundation Autism Research Initiative (SFARI) ASD gene database Although the contribution of individual candidates to autism will unlikely exceed an infinitesimal proportion of the burden, understanding each one can be of profound importance, especially for a given endophenotype [7].
The complexity of ASD can undoubtedly be attributed, in part, to both the extreme phenotypic heterogeneity and involvement of interactions between environmental stimuli and the genetic background. Our objective is to disentangle the genetic heterogeneity by developing biologically relevant sub-classifications of ASD [8]. Our underlying hypothesis is that etiologic heterogeneity is reflected in the observed clinical and genetic heterogeneity and that clarifying the clinical and genetic heterogeneity will resolve the etiologic heterogeneity. Previous phenotypic analysis [8] suggests the existence of two significant subgroups within the existing ASD classification.
It has been proposed that ASD results from the dysfunction of specific genetic pathways, an extension of an early "polygenic hypothesis". There appears to be a relationship between the degree of disruption of these pathways and the severity of ASD. Finer sub-classifications of phenotypes allow one to focus on pathways and convergence of specific phenotypes onto those pathways. One such pathway enrichment tool, PARIS, allows for pathway analysis by randomization incorporating structure [9]. Pathway analysis aggregates single variants results into a single score, allowing modest association signals to collectively reach significance, essentially increasing the signal to noise ratio.
One promising strategy is to focus analyses on modifiers of severity, descriptors of a particular trait such as repetitive behavior, social abilities in the context of family and genetic background. Focusing on a narrow effect may illuminate underlying genetic effects lost in the broader phenotype. Previous studies have used this approach for specific loci, but not genome-wide on a large familial dataset such as represented by the MSSNG data.
Here, we investigated the potential modifiers of severity via association with the Vineland Adaptive Behavior Scales (VABS). Scores on the Vineland Adaptive Behavior Scale, the most widely used measure of adaptive behavior in autism, can range from four standard deviations below the mean to more than two standard deviations above the mean in populations of ASD both with and without comorbid mental retardation [10]. The importance of adaptive behavior variability in autism is underscored by its strong contribution to prognosis [10]. Identifying sources of variability in adaptive behavior is critical to obtaining a more complete picture of development in autism as well as identification of treatment targets.
We conducted a large scale association analysis of the MSSNG whole genome sequencing data to elucidate potential modifiers of ASD severity. We have directly tested association by linear regression between 6,198,166 SNPs that passed quality control and Vineland Adaptive Behavior Scores. We verified our results through meta-analysis of two different sequencing platforms (Illumina and Complete Genomics) used in this dataset. Interestingly, the most significant variants direct us to a region that spans ~3Mb (chr3:47.5-50.5 Mb), (part of the biggest META-significant haplostretch of SNPs, n = 132) containing multiple variants on chromosome 3 including a nonsynonymous SNV, rs11539148, within the QARS gene (NM_001272073:c.A821G:p.N274S MAF = 0.0391) shown as Figure 1. In addition, other interesting loci within the region (chr3p21 enrichment 3.68e-11) direct us to RHOA, GNAI2, RBM6, RBM5, MON1A, SLC38A3, BSN, SEMA3F, TMEM89, CAMVK, FBXW12. Furthermore, the result has been replicated in the second batch of the same project set (n = 383) became available (total n = 956).
 Figure 1: Circos plot showing a META-significant haplostretch of SNPs, (n = 132) containing multiple variants on chromosome 3 including a highly interesting nonsynonymous SNV rs11539148 within QARS. (NM_001272073:c.A821G:p.N274S MAF = 0.0391). QARS is a Glutaminyl-TRNA Synthetase coding gene crucial in brain development. View Figure 1
Figure 1: Circos plot showing a META-significant haplostretch of SNPs, (n = 132) containing multiple variants on chromosome 3 including a highly interesting nonsynonymous SNV rs11539148 within QARS. (NM_001272073:c.A821G:p.N274S MAF = 0.0391). QARS is a Glutaminyl-TRNA Synthetase coding gene crucial in brain development. View Figure 1
QARS is a Glutaminyl-TRNA Synthetase is a protein coding gene associated with Microcephaly, Progressive, Seizures, And Cerebral And Cerebellar Atrophy and Microcephaly [11]. Among its related pathways are Viral mRNA Translation and Gene Expression. GO annotations related to this gene include nucleotide binding and ligase activity, forming aminoacyl-tRNA and related compounds. It plays a critical role in brain development and is expressed in multiple tissues. In this analysis, the minor allele is protective, given the genotypic specificity and VABS performance as shown in Figure 2A. The specific coding variant (rs115xxxx) is among the top ~0.1% of damaging variants as indicated by its CADD score (= X), a widely used scoring algorithm [12], to predict deleteriousness of single nucleotide variants.
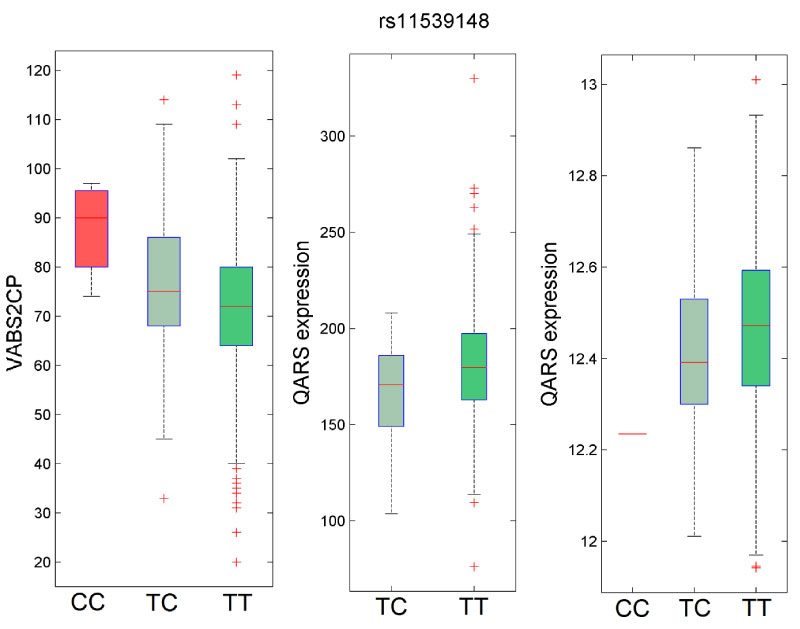 Figure 2: Effect of genotypes of rs11539148 on VABS2CP and QARS expression. A. protective effect B. QARS eQTLs across data suggest lower expression (p val -6) B C. eQTL analysis Boxplots for expression of QARS. Normalized gene expression (y-axis) is plotted by genotype in B. GTEX and C. Geuvadis. We observe a significantly lower expression. View Figure 2
Figure 2: Effect of genotypes of rs11539148 on VABS2CP and QARS expression. A. protective effect B. QARS eQTLs across data suggest lower expression (p val -6) B C. eQTL analysis Boxplots for expression of QARS. Normalized gene expression (y-axis) is plotted by genotype in B. GTEX and C. Geuvadis. We observe a significantly lower expression. View Figure 2
We also found rs11539148 to be a strong eQTL for QARS expression, as confirmed across three gene expression datasets including GTEX (2 tissues) Geuvadis (Figure 2B and Figure C). This variant is in a region that spans ~3Mb (chr3:47.5-50.5 Mb), with much higher density of significant SNPs around 49.8-50.2 Mb (Figure 1), significantly enriched region. Additionally, the LD structure is consistent across CEU and non-CEU populations as well as our MSSNG cohort.
A2M variant rs2228222 Haploreg v4.1 was used to investigate the regulatory potential, showing that rs276571 demonstrates a number of lines of evidence to support a function in disease causality, including mapping to an enhancer in B-lymphoblastoid cell lines, primary and T-regulatory cells. It also maps to a region of open chromatin, characterized by DNase hypersensitivity, shows evidence of binding of STAT3. Furthermore, analysis of a library of transcription factor binding site position weight matrices predicts that the SNP alters the binding site of two transcription factors. Furthermore, the locus has been shown to be a significant (0.000004) eQTL for IL20RA in GTEX.
Other variants in the region point to LYPD1, a member of the Lynx family of neurotransmitter receptor-binding proteins implicated in anxiety (ref); NCKAP5, previously implicated in autism (CNV, 2 cases; ref) and GPR39, a product of which has been implicated in depression (ref). Some other interesting loci include CACNA2D2 (P < 1 × 10−7, 16 markers) encoding a subunit of the voltage-dependent calcium channel complex and axon guidance receptor gene, DCC (P < 1 × 10−7). Calcium ion channel gene CACNA1C has been associated with Timothy syndrome [2]. Calcium channels mediate the entry of calcium ions into the cell upon membrane polarization. CACNA1C encodes the alpha-2/delta subunit of the voltage-dependent calcium channel complex. The complex consists of the main channel-forming subunit alpha-1, and auxiliary subunits alpha-2/delta, beta, and gamma. The auxiliary subunits function in the assembly and membrane localization of the complex and modulate calcium currents and channel activation/inactivation kinetics. The subunit encoded by this gene undergoes post-translational cleavage to yield the extracellular alpha2 peptide and a membrane-anchored delta polypeptide. This subunit is a receptor for the antiepileptic drug, gabapentin. Mutations in this gene are associated with early infantile epileptic encephalopathy. Single nucleotide polymorphisms in this gene are correlated with increased sensitivity to opioid drugs. Alternative splicing results in multiple transcript variants encoding different isoforms.
Pathway enrichment thus far in multiple settings has indicated that many known genes and loci involved in ASD risk converge into distinct biological processes: disruptions to synaptic functioning, chromatin remodelling, Wnt signalling, transcriptional regulation, interactions with FMR1 and, more broadly, MAPK signaling [13,14]. Hence, many such ASD associated variants converge onto common pathways. It is pertinent to ask whether the identified loci point to genes involved in common processes, active at specific cell types at the stages of development. We conducted a pathway enrichment analysis of the set of highly significant results (SNPs in the identified region as discussed) using tools such PARIS [9] and DAVID with primary results shown in Table 1. Interestingly, the significant pathways include axon guidance, circadian rhythm, addiction related pathways and vitamin B metabolism among others. Genes categorized a neurological, developmental and immune-related constitute 65% of all tested.
Moreover, the relationship of ASD detected genes with gene co-expression networks further points towards the importance of Wnt signaling and negative regulation of axon genesis among other network modules (Figure 3). To follow up these results, we extracted variants from contributing genes from significantly overrepresented categories to test how much variability in the VABS scores can be explained by the region. The variability explained in the single top pathway enrichment alone on affection status is 2% (P = 6.34 × 10−6). GCTA [15] REML was run on all SNPs from coding regions of multiple genes significant in PARIS to estimate the percentage variability explained by these gene sets. One such category is axon guidance (p < 0.001), with significant contributors being SEMA3B (inhibits axonal extension), SEMA4F ROBO2, SLIT1, NCK2, NRAS, PPP3CB, EPHB2, RHOA, DCC (Receptor for netrin required for axon guidance) (entire lists in SM). In the cocaine addiction interestingly one of the major contributors are the variants within GRIN2B Glutamate Ionotropic Receptor NMDA Type Subunit 2B also a contributor to other forms of addiction. Another category investigated whose strongest contributors are CUL1, CLOCK and PER is circadian rhythm which seems to be affected in ASD.
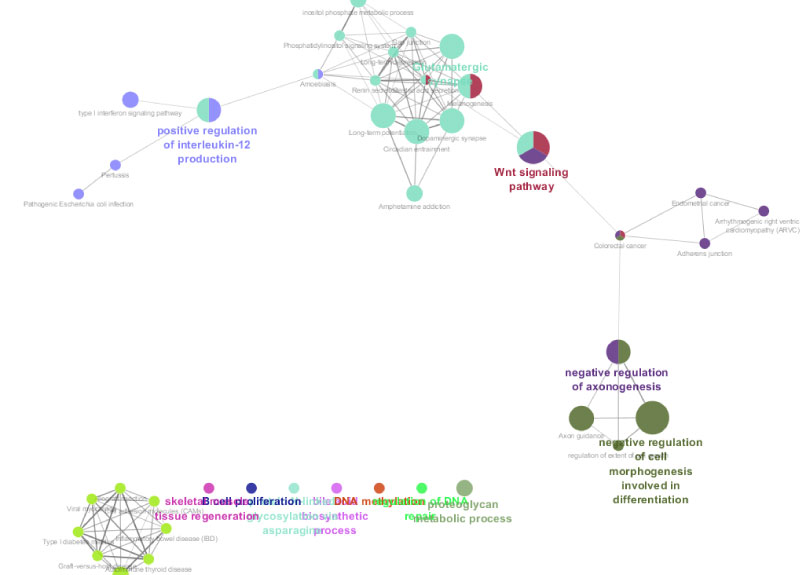 Figure 3: Network derived from pathway analysis of the significant region including QARS variant rs151. Individual modules are derived with ClueGO and based on gene expression data. View Figure 3
Figure 3: Network derived from pathway analysis of the significant region including QARS variant rs151. Individual modules are derived with ClueGO and based on gene expression data. View Figure 3
Other pathways include Wnt signaling 0.002, RHOA, CTBP2, TCF7L2 and interestingly repression of WNT target genes mediated by TCF7L2, TLE1 and CTBP2 (pval 0.000005). In terms of genes implicated in other disorders we get enrichment of 0.00004 of genes implicated in bipolar disorder such as CDK17, ZBTB18, FBXW12, ERC2, BSN, A2M and RHOA, A2M exonic variant rs2228222.
Addressing vexing questions posed by ASD heterogeneity via large scale pathway analysis hence leads to greater understanding of the biological mechanisms via genetic data and their translation into clinical uses. In addition, the significant gene set was inspected in terms of DGIdb to see if any of the genes in the region are found to interact with existing drugs. The 2 such implicated genes were CACNA2D2 Calcium Voltage-Gated Channel Auxiliary Subunit Alpha2delta 2 and GRIA1 Glutamate Receptor Ionotropic AMPA 1.
There is accumulating evidence ASD is caused by rare inherited or spontaneous genetic mutations, such as copy number changes and single nucleotide alterations. However, the genetic influences that have currently been found only account for about 15% of the cases…Here we detect a region that is a likely modifier of severity. It will be interesting to see the effects of producing recombinant QARS protein with a variant of interest + known variants that perturb tRNA synthesis. Given that lower expression is correlated with specific variant/genotype, in example we could perturb the tRNA in some other robust way, sh knock down. Interestingly, mutations in QARS have been reported as the causative variants in two unrelated families affected by progressive microcephaly [11]. Whole-exome sequencing of individuals from each family independently identified compound-heterozygous mutations in QARS as the only candidate causative variants [11]. Furthermore, aminoacylation activity of QARS was lower in cell lines carrying compound-heterozygous QARS mutations from affected individuals I-1 (A) and II-1 and II-2 (B) than in a normal control cell line. Single substitution in p.Gly45Val (I-3), p.Tyr57His (II-4), and p.Arg515Trp (II-3) also impaired QARS aminoacylation activity.
Autism is currently considered a disorder with multiple etiologies distributed across genetic/epigenetic environmental and behavioral domains which converge upon affected brain connectivity. The variability is the outcome of varying genetic liabilities coupled with environmental factors and molecular syndromes. One of the main issues likely preventing us from finding the missing links is the heterogeneity of the assessment tools for ASD clinical evaluations. Integrating the current genetic and clinical data from MSSNG and other cohorts into a unified platform will certainly lead to more effective translation of the results. Here we attempted one such largest thus far for overcoming the fragmentation of the data. Here we focused on VABS as it was a comprehensive measure of functioning well represented in the studied dataset. We were able to replicate the finding as see the effect of the minor allele upon, effect of consistent direction across several datasets. We furthermore observe smaller heads in the small number of cases we have with that measurement.
We envision that systematic study of all defined endophenotypes and underlying genomic pathways will yield important findings for susceptibility and understanding the pathogenesis of ASD within newly discovered subcategories. Our hypothesis is that a pathway approach disentangles the heterogeneity and direct us to the driver mechanisms of ASD. We looked at pathways as such perturbation model encompassing vulnerabilities to pleiotropic genes associated with important molecular mechanisms, their interactions with environmental input and effects upon multiple systemic comorbidities is likely to direct us to the mechanisms underlying each of the subtypes. We aimed to look for associations with VABS as such can point to specific modifiers of severity, and furthermore likely endophenotypes. We furthermore inspect the significant and identified region for pathway enrichment and show the result of such genetic risk scores. We examined pathway results in more detail to determine if there are specific subsets of "driver genes" that are generating most of the significance. In congruence with our pathways findings SNPs which overlap genes in common molecular pathways, such as calcium channel signaling, are shared in ASD, attention deficit-hyperactivity disorder, bipolar disorder, major depressive disorder, and schizophrenia. However, the sample size for the sub-classifications is small and further investigations of this GRS based on larger, well characterized samples of patients are clearly warranted. We used the present findings to confirm/refine preliminary results of autism subcategories that were detected using genetic and phenotypic data so far. We furthermore confirmed as significant difference between the clusters on severity using the variants in the significant and discussed region.
We detect a protective variant within a region of significance that is implicated in the severity of ASD. Whereas aetiological heterogeneity, variable penetrance and genetic pleiotropy are pervasive characteristics of autism genetics we show that through careful analysis and focus on outcomes, we can detect interesting and replicable results of genetic nature that would be undetected otherwise. As genetic predisposition may be different for almost every ASD individual, understanding the common mechanisms for endophenotypes may help elucidate ASD causal mechanisms. It does correlate with lower expression level and furthermore in a separate set, has effects upon head circumference. The effect is highly significant and reproducible in another dataset. Converging biological insights derived from genetic studies are warranting to reveal potential targets for the development of pharmacological compounds. We envision that careful studies of endophenotypes will contribute to such and more studies but foremost to the understanding of abberant mechanisms involved in development of ASD.