Introduction: There is a significant imbalance in the generation of NAD/NADH+ (niacin), which affects chemical reactions in the intracellular environment in COVID-19 and yellow fever. From tryptophan and its metabolic pathways and oxidative stress, the process of understanding SARS-CoV-2 infection becomes more concrete, as the infection seems to interfere in these metabolic pathways, in addition to the paradoxical role of kynurenine that causes inflammation by blocking the BH4 pathway. Understanding metabolic changes in the elderly, people with type 2 diabetes (DM2), obese people or other chronic diseases with an inflammatory profile is to understand the severity of COVID-19 to improve clinical management.
Hypothesis: SARS-CoV-2 may control the human immune response by acting on Furins and Cathepsins or triggering significant hypoxia; promotes the internalization of ACE-2, resulting in low absorption of some amino acids in the intestine, triggering immune suppression and metabolic syndrome (MS).
Results: From overweight people to people with higher Body Mass Index (BMI) or insulin resistance, there is a tendency to hyperthermia by thermogenesis due to the consumption of serotonin (5-HT) and norepinephrine (NOR) in fat cells, triggering an increased inflammatory state and cell damage. It is typical of critically ill patients with COVID-19 who have been treated with antipyretic and antimicrobial drugs at elevated temperatures, but the correct treatment is an insulin pump. It is not fever; it is hyperthermia. The state of inflammation is related to the Kynurenine/BH4 imbalance.
Objectives: This article aims to raise doubts to generate more discussions and bring more substrates to improve the patient's COVID-19. The primary pathophysiology of COVID-1 may be tryptophan syndrome due to kynurenine/BH4 imbalance and the maintenance of the hypoxic environment that causes immunosuppression, tolerance and inflammation due to oxidative stress. A signature of innate immunity, oxidative stress, and tryptophan pathway imbalance.
As a ripening of the first [1] article we published, this new manuscript intends to explain, in a more organized way, the dependence relationship of COVID-19 with the concentrations of NAD/NADH+ and why sustaining the patient in a hypoxic environment is causing immunosuppression, pulmonary fibrosis and more consequences that will delimit prognosis and the ability to live depending on the pulmonary sequelae.
aA prelude is a piece of music that precedes "something else", a fugue, for instance, or it could be the opening or the introduction of a suite. It normally has the function of preparing the listeners' ears for a certain affect through the composer's choice of key and time signature.Based on the findings of Pelletier, et al. [2] and with a previous article published on severity predictors in Yellow Fever disease, in which one of the predictor of severity is neutrophilia [1], reinforces the idea contained in the first article published by Zanella and Galvão [3] that the internalization of ACE-2 generates a lack of Try and Phe, whose results are large changes in aerobic respiration due to a deficit in NAD/NADH + and in the serotoninergic and dopaminergic pathways. The change in Try metabolism tends to produce Kynurenine (KYN) to control inflammation, since Kynurenine is immunosuppressive.
KYN binds to the aryl hydrocarbon receptor (AhR) although it appears to act importantly on neutrophils, preventing the formation of ROS and causing Treg/Th17 imbalance making the medium more tolerogenic. AhR is expressed in B cells, helper T cells 17, regulatory T cells, thymocytes and monocyte-derived cells (macrophages and dendritic cells), negatively regulating dendritic cells, and may be affected by bacterial kynurenine. It was previously shown that Pseudomonas aeruginosa uses KYN production to evade the host's immune response.
At the beginning of the pandemic, when we restructured the Hospital wards to receive patient COVID-19, some publications reported that patients with Parkinson's Disease had worsening symptoms when infected with SARS-CoV-2. At the same time, neurological symptoms during acute infection or after discharge from the ICU were frequent, and in most cases, there was no ischemic injury or any other injury after computed tomography or magnetic resonance imaging. Given these facts, I started from the hypothesis that the dopaminergic and serotoninergic pathways could be impaired in COVID-19, resulting in a first review article. Today, much evidence after metabolomics and gene expression experiments seems to support the hypothesis that in COVID-19, we have a mainly Tryptophan (Try) and Phenylalanine (Phe) deficiency syndrome.
This article uses the acute infection model in Yellow Fever to exemplify the immune response to acute infection in COVID-19 since both diseases progress similarly: viraemic phase, defervescence phase and inflammatory/toxaemic convalescent phase. For the chronic phase of COVID-19, I use models in which tryptophan and the immune response are influenced by chronic inflammatory imbalance, which occurs in "Long COVID-19 syndrome".
"Long COVID-19 Syndrome" is a phase with an inflammatory profile and tolerance variables dependent on comorbidities, genetic variability, time of exposure to hypoxemia that led the patient to the severity of the disease, associated with lipids - glycaemic dysmetabolism, which may reappear in thromboembolic events. In summary, a thrombo-metabolic and immunosuppressive syndrome triggered by SARS-COV-2 (TMISy-CoV-2) infection. A chronic phase that is still poorly diagnosed has been a cause of high mortality after the acute phase of the disease, which can occur soon after the end of the acute phase or appear only or several times later. It is an unrecognized and underdiagnosed phase.
On the metabolic pathways of tryptophan (Try) and phenylalanine (Phe) in yellow fever and COVID-19.
In the metabolic pathways of tryptophan (Try) and phenylalanine (Phe) in yellow fever and COVID-19.
Try and Phe are neutral amino acids absorbed in the intestine by the gene-encoded transporter and identified as B0AT1 [4-7]. This transporter is responsible for the function of the B0 system in the intestine and kidneys. The protein is expressed exclusively in the brush border membrane of intestinal and renal epithelial cells. It is a Na+-coupled transporter for neutral amino acids, and defects in this transport system explain the hyperexcretion of neutral amino acids in urine in patients with protein collectrin associated with Hartnup's disease (kidney) or ACE2 (intestinal). In yellow fever (Figure 1), there is no change in amino acid absorption mediated by yellow fever virus infection. In COVID-19, ACE-2 is internalized through SARS-COV-2 infection, the consequence of which will be Try and Phe malabsorption in intestinal cells.
Try has three possible pathways for its metabolism according to the enzymatic pathway. There are the following enzymes IDO1 enzyme, IDO2 enzyme, TDO enzyme, tryptophan hydroxylase 1 and 2 (TPH1 enzyme and TPH2 enzyme) that allow 3 possible vias: 1) Kynurenine pathway; 2) Serotonin (5-HT) pathway; and 3) Vitamin B3 pathway (NAD/NADH+). When in homeostasis, Try is metabolized by TDO produced in the liver. Of the absorbed tryptophan, about 95% goes to the peripheral pathway (serum) and only 5% to the Central Nervous System (CNS) to generate 5-HT and melatonin. About 95% of serotonin is synthesized and stored in the gastrointestinal tract, where it acts as a paracrine messenger to modulate sensation, secretion, and motility, and is also involved in appetite control [8,9].
Try enzymes indoleamine-2,3-dioxygenase (IDO) and tryptophan-2,3-dioxygenase (TDO or TDO2), regulate the first and rate-limiting step of the Kyn pathway TDO supports up to 95% of hepatic Trp metabolism. Moreover, TDO can be detected in the kidney, skin, and other tissues, including the placenta, pregnant uterus, epididymis, testis, and brain after being stimulated. Inflammation (mediated by infection or not) and hepatic hypoxia shift the Try metabolism to be carried out by the enzyme IDO, as these conditions decrease the expression of TDO in the liver and increase the expression of IDO in macrophages and dendritic cells (DCs), connective tissue (fibroblast) and epithelial tissue (pulmonary, renal, gastrointestinal and vascular), mainly stimulated by interferon-gamma (IFN-γ) and other lipid mediators such as prostaglandin E2 (PGE2) and pathogen particles such as lipopolysaccharide (LPS). IDO-1 is the predominant of the two enzymes and is found in many cell types including, but not limited to, astrocytes, neurons, microglia, dendritic cells, monocytes, and macrophage, whereas IDO-2 has only been found in a smaller subset of cells, primarily dendritic and stem cells and some cancer lines. IDO-1/IDO-2 is the enzyme responsible for catalysing the rate-limiting step in the peripheral tissues and depends on the active form of superoxide (O2-) [9-12] (Figure 1 and Figure 2).
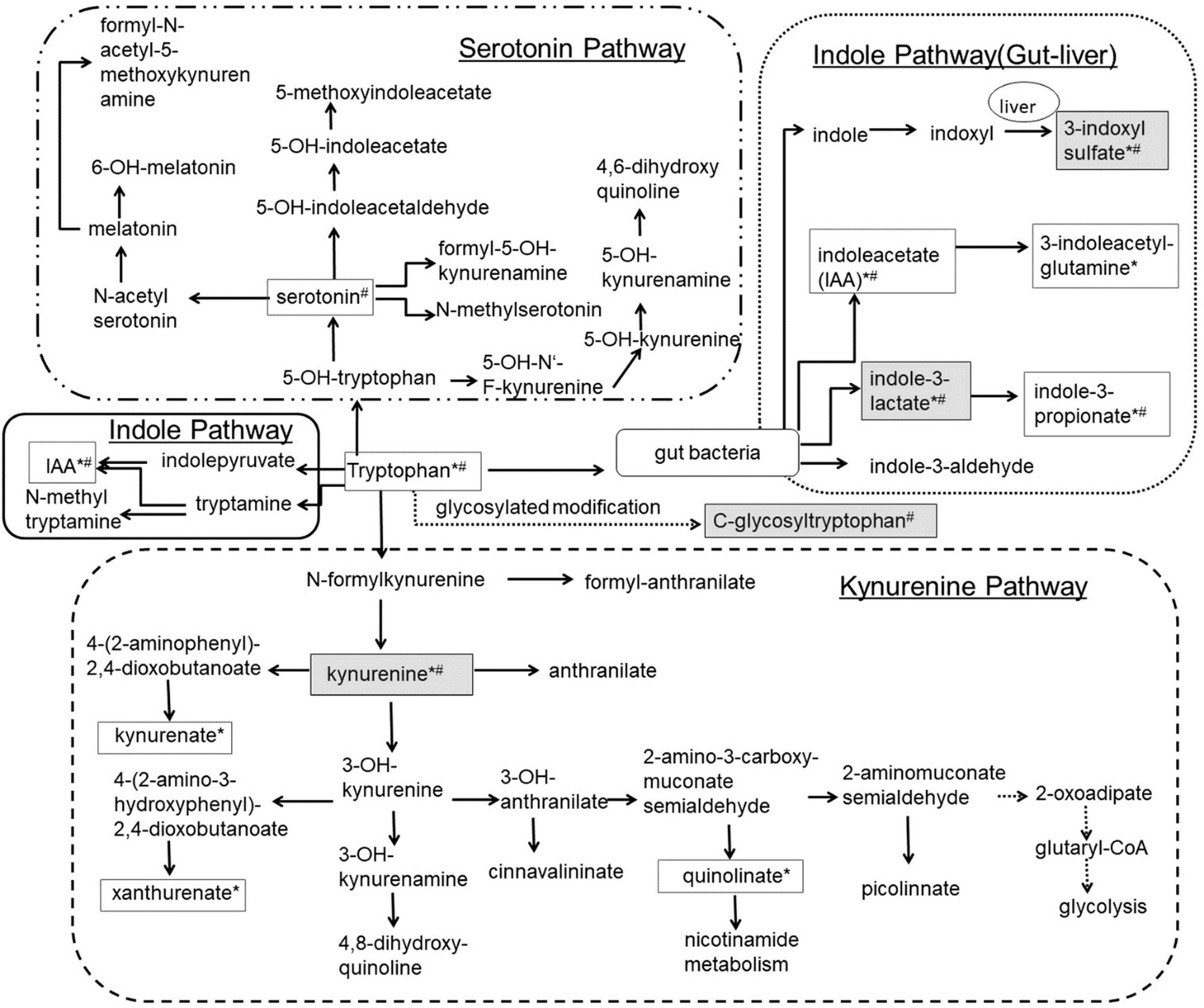 Figure 1: L-Tryptophan (L-Try) is an essential amino acid required for protein biosynthesis. It is also a biochemical precursor of metabolites that significantly affect mammalian physiology, including gastrointestinal functions, immunity, metabolism, and the nervous system. In the gastrointestinal tract L-Try metabolism can follow three significant pathways, all of which are influenced by the gut microbiota: (i) The kynurenine pathway (KP) in both immune and epithelial cells, (ii) The serotonin (5-hydroxytryptamine, 5-HT;production pathway in enterochromaffin cells (ECCs), a specialized subtype of intestinal epithelial cell, and (iii) Direct transformation by the gut microbiota of L-Try into several molecules, including ligands of the aryl hydrocarbon receptor (AhR). In mammalian cells, most L-Trp is metabolized via the KP, while the remainder is utilized in the synthesis of 5-HT and melatonin (MT). In KP, L-Trp is catabolized into the unstable derivative N-formyl-L-kynurenine (NFK) by rate-limiting enzymes tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenases (IDO1/IDO2). Globally, the enzymes of KP are expressed in a tissue-specific manner. TDO is expressed in the liver, whereas IDO1 is expressed in many cell types and tissues and is inducible by cytokines NFK is rapidly metabolized by kynurenine formamidase (expressed in liver, kidney, and brain) to form L-kynurenine (L-kyn). L-kyn is a crucial metabolite with potent immunoregulatory functions through its binding to AhR. L-kyn is mainly metabolized by kynurenine monooxygenase (KMO) to form 3-hydroxykynurenine (3-HK). 3-HK is then degraded to 3-hydroxyanthranilic acid (3-HAA) by kynureninase (KYNU). KYNU is subsequently metabolized to 2-amino-3-carboxymuconic 6-semialdehyde (ACMS) by 3-hydroxyanthranilic acid 3,4-dioxygenase (3-HAO). The former is expressed in the liver, kidney, central nervous system (CNS), and placenta, while the latter has a broad tissue distribution. ACMS can be cyclized to quinolinic acid (QUIN) or metabolized by the enzyme 2-amino-3-carboxymuconate-semialdehyde decarboxylase (ACMSD), found mainly in the kidney and to a lesser extent in the liver, and responsible for the synthesis of 2-aminomuconic-6-semialdehyde (AMS). AMS is either metabolized by 2-aminomuconic semialdehyde dehydrogenase (AMSD) to result in acetyl-CoA or cyclized nonenzymatically to form picolinic acid (PICA). In the CNS, QUIN is mainly produced by microglia. It acts as a neurotoxic agent on astrocytes mainly by its selective agonist effect on ionotropic glutamate glutamatergic N-methyl-D-aspartate (NMDA) receptors. However, mechanisms determining the engagement of KP in its synthesis remain undetermined. QUIN is also a precursor for the de novo synthesis pathway for NAD via the enzyme Quinolinate phosphoribosyltransferase (QPRT) expressed mainly in the liver and kidney. NAD is a cofactor for numerous enzymes involved in cellular energy metabolism, adaptive responses of cells to bioenergetic and oxidative stress, and genome stability. Its deficiency affects tissues that need high cellular energy, such as the brain, gut, and skin, causing pellagra. Finally, PICA is a neuroprotective molecule whose concentration is reduced in the serum of patients with autism, and plasma and cerebrospinal fluid (CSF) of subjects who have attempted suicide [143].
View Figure 1
Figure 1: L-Tryptophan (L-Try) is an essential amino acid required for protein biosynthesis. It is also a biochemical precursor of metabolites that significantly affect mammalian physiology, including gastrointestinal functions, immunity, metabolism, and the nervous system. In the gastrointestinal tract L-Try metabolism can follow three significant pathways, all of which are influenced by the gut microbiota: (i) The kynurenine pathway (KP) in both immune and epithelial cells, (ii) The serotonin (5-hydroxytryptamine, 5-HT;production pathway in enterochromaffin cells (ECCs), a specialized subtype of intestinal epithelial cell, and (iii) Direct transformation by the gut microbiota of L-Try into several molecules, including ligands of the aryl hydrocarbon receptor (AhR). In mammalian cells, most L-Trp is metabolized via the KP, while the remainder is utilized in the synthesis of 5-HT and melatonin (MT). In KP, L-Trp is catabolized into the unstable derivative N-formyl-L-kynurenine (NFK) by rate-limiting enzymes tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenases (IDO1/IDO2). Globally, the enzymes of KP are expressed in a tissue-specific manner. TDO is expressed in the liver, whereas IDO1 is expressed in many cell types and tissues and is inducible by cytokines NFK is rapidly metabolized by kynurenine formamidase (expressed in liver, kidney, and brain) to form L-kynurenine (L-kyn). L-kyn is a crucial metabolite with potent immunoregulatory functions through its binding to AhR. L-kyn is mainly metabolized by kynurenine monooxygenase (KMO) to form 3-hydroxykynurenine (3-HK). 3-HK is then degraded to 3-hydroxyanthranilic acid (3-HAA) by kynureninase (KYNU). KYNU is subsequently metabolized to 2-amino-3-carboxymuconic 6-semialdehyde (ACMS) by 3-hydroxyanthranilic acid 3,4-dioxygenase (3-HAO). The former is expressed in the liver, kidney, central nervous system (CNS), and placenta, while the latter has a broad tissue distribution. ACMS can be cyclized to quinolinic acid (QUIN) or metabolized by the enzyme 2-amino-3-carboxymuconate-semialdehyde decarboxylase (ACMSD), found mainly in the kidney and to a lesser extent in the liver, and responsible for the synthesis of 2-aminomuconic-6-semialdehyde (AMS). AMS is either metabolized by 2-aminomuconic semialdehyde dehydrogenase (AMSD) to result in acetyl-CoA or cyclized nonenzymatically to form picolinic acid (PICA). In the CNS, QUIN is mainly produced by microglia. It acts as a neurotoxic agent on astrocytes mainly by its selective agonist effect on ionotropic glutamate glutamatergic N-methyl-D-aspartate (NMDA) receptors. However, mechanisms determining the engagement of KP in its synthesis remain undetermined. QUIN is also a precursor for the de novo synthesis pathway for NAD via the enzyme Quinolinate phosphoribosyltransferase (QPRT) expressed mainly in the liver and kidney. NAD is a cofactor for numerous enzymes involved in cellular energy metabolism, adaptive responses of cells to bioenergetic and oxidative stress, and genome stability. Its deficiency affects tissues that need high cellular energy, such as the brain, gut, and skin, causing pellagra. Finally, PICA is a neuroprotective molecule whose concentration is reduced in the serum of patients with autism, and plasma and cerebrospinal fluid (CSF) of subjects who have attempted suicide [143].
View Figure 1
 Figure 2: Mechanisms involved the regulation of inflammation by Try metabolism. Inflammation activates Trp metabolism and causes systemic and intra- and extra-cellular changes in the Kyn/Try ratio that suppress the inflammatory response (A). The molecular steps involved in the immunomodulatory effect of activation of Try metabolism (B): An inflammatory stimulus activates IDO (and in specific instances TDO) in immune and non-immune cells causing reduced Try systemic and local Try levels and increased intra- and extracellular Kyn content (1); inflammation induces increased expression of AhR (2) that is activated by its ligand Kyn and results in the secretion of anti-inflammatory cytokines such as IL-10 (3); AhR ligand-activation causes phosphorylation of IDO and results in sustained IDO activity and the secretion of TGF-β, which is involved in a feedback loop by inducing IDO phosphorylation (4); inflammatory cytokines such as TGF-β and IL-10 induce the amino acid transporter SLC7A5 on the plasma membrane of naive T-cells causing transport of Kyn into the T cell (5); activation of GCN2 by Try depletion and AhR ligand-activation by Kyn cause the differentiation of naïve T cells toward regulatory T cells (6).Changes in kynurenine pathway during chronic kidney disease. In chronic kidney disease was found lower serum concentration of Trp in both, animal and human studies. Conversely, higher serum KYN, KYNA, 3-OHKYN, AA, XA and QA were reported, suggesting kynurenine pathway activation in chronic kidney disease. Acute kidney injury (AKI) following ischemia-reperfusion injury (IRI) has a high mortality and lacks specific therapies. Here, we report that mice lacking kynurenine 3-monooxygenase (KMO) activity (Kmonull mice) are protected against AKI after renal IRI. This advances our previous work showing that KMO blockade protects against acute lung injury and AKI in experimental multiple organ failure caused by acute pancreatitis. We show that KMO is highly expressed in the kidney and exerts major metabolic control over the biologically-active kynurenine metabolites 3-hydroxykynurenine, kynurenic acid and downstream metabolites. In experimental AKI induced by unilateral kidney IRI, Kmonull mice had preserved renal function, reduced renal tubular cell injury, and fewer infiltrating neutrophils compared to wildtype (Kmowt) control mice. Together, these data confirm that flux through KMO contributes to AKI after IRI and supports the rationale for KMO inhibition as a therapeutic strategy to protect against AKI during critical illness. 3-OHKYN 3-hydroxykynurenine, AA anthranilic acid, IDO indoleamine 2,3-dioxygenase, KAT kynurenine aminotransferase, KYN kynurenine, KYNA kynurenic acid, QA quinolinic acid, TDO tryptophan 2,3-dioxygenase, Try tryptophan, XA xanthurenic acid [167,168].
View Figure 2
Figure 2: Mechanisms involved the regulation of inflammation by Try metabolism. Inflammation activates Trp metabolism and causes systemic and intra- and extra-cellular changes in the Kyn/Try ratio that suppress the inflammatory response (A). The molecular steps involved in the immunomodulatory effect of activation of Try metabolism (B): An inflammatory stimulus activates IDO (and in specific instances TDO) in immune and non-immune cells causing reduced Try systemic and local Try levels and increased intra- and extracellular Kyn content (1); inflammation induces increased expression of AhR (2) that is activated by its ligand Kyn and results in the secretion of anti-inflammatory cytokines such as IL-10 (3); AhR ligand-activation causes phosphorylation of IDO and results in sustained IDO activity and the secretion of TGF-β, which is involved in a feedback loop by inducing IDO phosphorylation (4); inflammatory cytokines such as TGF-β and IL-10 induce the amino acid transporter SLC7A5 on the plasma membrane of naive T-cells causing transport of Kyn into the T cell (5); activation of GCN2 by Try depletion and AhR ligand-activation by Kyn cause the differentiation of naïve T cells toward regulatory T cells (6).Changes in kynurenine pathway during chronic kidney disease. In chronic kidney disease was found lower serum concentration of Trp in both, animal and human studies. Conversely, higher serum KYN, KYNA, 3-OHKYN, AA, XA and QA were reported, suggesting kynurenine pathway activation in chronic kidney disease. Acute kidney injury (AKI) following ischemia-reperfusion injury (IRI) has a high mortality and lacks specific therapies. Here, we report that mice lacking kynurenine 3-monooxygenase (KMO) activity (Kmonull mice) are protected against AKI after renal IRI. This advances our previous work showing that KMO blockade protects against acute lung injury and AKI in experimental multiple organ failure caused by acute pancreatitis. We show that KMO is highly expressed in the kidney and exerts major metabolic control over the biologically-active kynurenine metabolites 3-hydroxykynurenine, kynurenic acid and downstream metabolites. In experimental AKI induced by unilateral kidney IRI, Kmonull mice had preserved renal function, reduced renal tubular cell injury, and fewer infiltrating neutrophils compared to wildtype (Kmowt) control mice. Together, these data confirm that flux through KMO contributes to AKI after IRI and supports the rationale for KMO inhibition as a therapeutic strategy to protect against AKI during critical illness. 3-OHKYN 3-hydroxykynurenine, AA anthranilic acid, IDO indoleamine 2,3-dioxygenase, KAT kynurenine aminotransferase, KYN kynurenine, KYNA kynurenic acid, QA quinolinic acid, TDO tryptophan 2,3-dioxygenase, Try tryptophan, XA xanthurenic acid [167,168].
View Figure 2
The Kynurenine pathway (Figure 2) is initiated by conversion of l-tryptophan, by either of the enzymes tryptophan-2,3-dioxygenase or indoleamine 2,3-dioxygenase each forming formyl-kynurenine, which is then further degraded to kynurenine, the precursor of several bioactive compounds, including kynurenic acid, quinolinic acid, picolinic acid, and 3-hydroxyanthranilic acid. The pathway is responsible for over 90% of tryptophan metabolism in the periphery.
Kynureninase is a pyridoxal phosphate-dependent enzyme inhibited by oestrogen and metabolites, with both vitamin B6 deficiency and inhibition. There is an increase in Kyn, HK, XA, and HaA and a decrease in KA and AA because of the simulation of a general vitamin B-6 deficiency in which the activities of the enzymes were not completely reduced [8,13].
Naive CD4+T cells can differentiate into T helper (Th) cells, Th1, Th2, and Th17 lineages and regulatory T (Treg or CD3+CD4+FOXP3+) cells. -6 and transforming growth factor β (TGFβ) is considered critical for the differentiation into Th17 cells. IL-21 and IL-23 pathways are involved in IL-6-programmed Th-17 cell differentiation. Various cytokines regulate th17 differentiation. TGF-β and IL-6 induced th17 differentiation in mice, and IL-1β but not TGF-β has been shown to participate in the development of Th17 cells together with IL-6 in humans.
The development of Th17 cells is negatively regulated by IFN-γ, IL-27, and IL-2, the signals of which are dependent on Stat1 (IFN-γ and IL-27) and Stat5 (IL-2), respectively. The orphan nuclear receptors, retinoid-related orphan receptor γ (RORγ) and RORα have been identified as the key transcription factors that determine the differentiation of Th17 lineage. AhR is involved in the differentiation of Th17 cells by regulating Stat1 activation, which suppresses Th17 cell differentiation under Th17-polarizing conditions (Figure 1 and Figure 2).
IL-27 and IFN-γ suppressed the generation of Th17 cells without significant effects on the expression of RORγ. Th17 differentiation is positively regulated by IL-6 or IL-21 in combination with TGF-β and negatively regulated by IFN-γ or IL -27, which Stat3 and Stat1, respectively control. When the TGF-β1 signalling pathway is activated, the expression of RORγt and Foxp3 is upregulated. Whether naïve T cells polarize to a Th17 phenotype, or a regulatory phenotype largely depends on the surrounding microenvironments. Foxp3 can inhibit Th17 development by directly binding to RORγt. Without IL-6, the TGF-β signalling pathway reinforces this inhibition and favours the formation of Treg from naïve T cells. In the presence of IL-6, STAT3 can be activated, and Foxp3 is released from RORγt. During differentiation of Th17, the expression of IL-23R is also upregulated, and activated IL-23R can induce Th17 differentiation. IL-23R is also very important for the proliferation and maintenance of the phenotype of Th17 cells after differentiation. The IL-23 signalling pathway can also activate STAT3 and inhibit IL-10 production. TGF-β1-induced Th17 differentiation can occur without IL-6 if there is sufficient IL-21 [14-20] (Figure 3).
 Figure 3: Catastrophic Treg x T17 imbalance in SARS-CoV-2 infection and Tryptophan starvation. SARS-CoV-2 infection by ACE-2 internalization and virus capacity of inhibiting. SARS-CoV-2 infection occurs internalization of ACE-2 and inhibition of INF-gamma by viral action. These are the first forms of polarization for IL-6, diabetic, obese, and older adults, and other diseases present this polarization more accentuated because their comorbidities frequently inflame them. SARS-CoV-2 is a disease that promotes angiogenesis and enormous tolerance due to the TH17 and Treg imbalance.
On the one hand, we have the infection promoting a tendency towards TH1 but inhibited by INF-gamma regulation. The lymphocytes tend towards Treg, and this TCD4 + Th2 function establishes a negative relationship in the control of the Th1 pathway. Thus NK, Th1, TCD8 + initiate a process of apoptosis and downregulation of its active pathways in inflammation. The large production of ADO1 accentuates this pathway by macrophages and dendritic cells, so the Tryptophan pathway is aimed at inhibition, that is, tolerance. The high consumption of 5-HT by the angiogenic process depletes serotonin, bypassing the tryptophan pathway to produce this essential molecule in angiogenesis and the development of tumours; it is a vasodilator stimulus. Lack of Tryptophan in the tolerogenic Indoleamine 2,3-dioxygenase (ADO)1 pathway polarizes the immune system to Treg. The significant lack of tryptophan promotes the consumptive syndrome by sarcopenia and the lack of vitamin B3, shifting the metabolism towards anaerobic respiration due to the lack of NAD/NADH+. NAD+ has a positive role in polarizing Treg to Th17, but it is made impossible by the anaerobic pathway. This process triggers a metabolic explosion with high apoptosis that is self-fuelled by the production of IL-6 via Th2 and by monocytes and macrophages stimulated continuously by new tissue lesions or new bacterial infections, mainly by gram negatives that stimulate IFN-γ. In this hurricane, even more exacerbated, the constant pro-inflammatory stimulus sustains an established tolerogenic state.
Thus, constant inflammation vis-a-vis a state of tolerance; there is a high probability of autoimmune diseases. Also, the toxic metabolites of tryptophan produce many neurological symptoms. Concerning the vessels, the continuous process of aggregation and reconstruction facilitates thrombosis mechanisms, and the high expression of adhesins associated with the demand for 5-HT is highly chemotactic for eosinophils. Eosinophils pass through all tissues so that a significant systemic hypereosinophilic syndrome mediated by serotonin is present, IL-6 activating mast cells and basophils. Finally, all types of eosinophilic vasculitis can happen in these patients. The intensity of all this complex pathophysiology depends on the inflammatory diseases that the patient has as comorbidities and on the vascular injury caused by the viral infection, so one should never avoid intubation in a patient who is on the threshold of the PaO2/FiO2 ratio. Not intubating is damaging more endothelium and pushing the metabolic and immune uncontrolled explained here. The centre of the Figure shows that perhaps some eosinophilia or many tissue eosinophilia depends on how the interaction occurs between cells and cytokines. It is essential to note the different orbits of neutrophils responsible, above all, for the formation of NETs and are significant links to platelet activation. Serotonin (5-HT) neutrophils follow an orbit and separate, as their stimulus, in this pathophysiology, is mainly influenced by autoantibodies of the ANCA type, although others may also appear and be involved. The design in the form of orbits intends to show that PMNs influence each other depending on the distance and their alignment. This characteristic, with the specificities of each patient, and hypoxemia will be the conductor who orchestrates the signs and symptoms. The cause of variable eosinophilia and the unique orbit of neutrophils appears to be linked to the problematic metabolism of tryptophan, whose shift to Kynurenine may be the cause of cyclic neutrophilia [2].
View Figure 3
Figure 3: Catastrophic Treg x T17 imbalance in SARS-CoV-2 infection and Tryptophan starvation. SARS-CoV-2 infection by ACE-2 internalization and virus capacity of inhibiting. SARS-CoV-2 infection occurs internalization of ACE-2 and inhibition of INF-gamma by viral action. These are the first forms of polarization for IL-6, diabetic, obese, and older adults, and other diseases present this polarization more accentuated because their comorbidities frequently inflame them. SARS-CoV-2 is a disease that promotes angiogenesis and enormous tolerance due to the TH17 and Treg imbalance.
On the one hand, we have the infection promoting a tendency towards TH1 but inhibited by INF-gamma regulation. The lymphocytes tend towards Treg, and this TCD4 + Th2 function establishes a negative relationship in the control of the Th1 pathway. Thus NK, Th1, TCD8 + initiate a process of apoptosis and downregulation of its active pathways in inflammation. The large production of ADO1 accentuates this pathway by macrophages and dendritic cells, so the Tryptophan pathway is aimed at inhibition, that is, tolerance. The high consumption of 5-HT by the angiogenic process depletes serotonin, bypassing the tryptophan pathway to produce this essential molecule in angiogenesis and the development of tumours; it is a vasodilator stimulus. Lack of Tryptophan in the tolerogenic Indoleamine 2,3-dioxygenase (ADO)1 pathway polarizes the immune system to Treg. The significant lack of tryptophan promotes the consumptive syndrome by sarcopenia and the lack of vitamin B3, shifting the metabolism towards anaerobic respiration due to the lack of NAD/NADH+. NAD+ has a positive role in polarizing Treg to Th17, but it is made impossible by the anaerobic pathway. This process triggers a metabolic explosion with high apoptosis that is self-fuelled by the production of IL-6 via Th2 and by monocytes and macrophages stimulated continuously by new tissue lesions or new bacterial infections, mainly by gram negatives that stimulate IFN-γ. In this hurricane, even more exacerbated, the constant pro-inflammatory stimulus sustains an established tolerogenic state.
Thus, constant inflammation vis-a-vis a state of tolerance; there is a high probability of autoimmune diseases. Also, the toxic metabolites of tryptophan produce many neurological symptoms. Concerning the vessels, the continuous process of aggregation and reconstruction facilitates thrombosis mechanisms, and the high expression of adhesins associated with the demand for 5-HT is highly chemotactic for eosinophils. Eosinophils pass through all tissues so that a significant systemic hypereosinophilic syndrome mediated by serotonin is present, IL-6 activating mast cells and basophils. Finally, all types of eosinophilic vasculitis can happen in these patients. The intensity of all this complex pathophysiology depends on the inflammatory diseases that the patient has as comorbidities and on the vascular injury caused by the viral infection, so one should never avoid intubation in a patient who is on the threshold of the PaO2/FiO2 ratio. Not intubating is damaging more endothelium and pushing the metabolic and immune uncontrolled explained here. The centre of the Figure shows that perhaps some eosinophilia or many tissue eosinophilia depends on how the interaction occurs between cells and cytokines. It is essential to note the different orbits of neutrophils responsible, above all, for the formation of NETs and are significant links to platelet activation. Serotonin (5-HT) neutrophils follow an orbit and separate, as their stimulus, in this pathophysiology, is mainly influenced by autoantibodies of the ANCA type, although others may also appear and be involved. The design in the form of orbits intends to show that PMNs influence each other depending on the distance and their alignment. This characteristic, with the specificities of each patient, and hypoxemia will be the conductor who orchestrates the signs and symptoms. The cause of variable eosinophilia and the unique orbit of neutrophils appears to be linked to the problematic metabolism of tryptophan, whose shift to Kynurenine may be the cause of cyclic neutrophilia [2].
View Figure 3
Aryl hydrocarbon receptor (AhR) is a member of the basic helix-loop-helix-(bHLH) superfamily of transcription factors associated with cellular responses to environmental stimuli, such as xenobiotics and oxygen levels.
AHR is expressed in the innate immune system cells, such as dendritic cells, macrophages, natural killer cells, and lymphoid tissue inducer-like cells. In contrast to other danger-sensing systems, such as the TLR pathways, AHR signals are thought to convey intrinsic metabolic or oxidative stress in a cell type-specific manner. Engagement of AHR with different ligands modulates the expression of surface molecules of dendritic cells and the secretion of cytokines with either proinflammatory or tolerogenic net effects [21,22].
Aryl hydrocarbon receptor (AhR) is a member of the basic helix-loop-helix-(bHLH) superfamily of transcription factors associated with cellular responses to environmental stimuli, such as xenobiotics and oxygen levels. AHR is expressed in the innate immune system cells, such as dendritic cells, macrophages, natural killer cells, and lymphoid tissue inducer-like cells. In contrast to other danger-sensing systems, such as the TLR pathways, AHR signals are thought to convey intrinsic metabolic or oxidative stress in a cell-type-specific manner. Engagement of AHR with different ligands modulates the expression of surface molecules of dendritic cells and the secretion of cytokines with either proinflammatory or tolerogenic effects.
The cytokine profile in elderly patients has an immune signature associated with the disease with elevated CXCL8, IL-10, IL-15, IL-27, and TNF-α positively correlates with older age, more extended hospitalization, and a more severe form of the disease and may thus represent the leading signature in critical COVID-19 patients [23].
In the remaining patients, those with the severity of plasma concentrations of IFN-α, IP-10, MIG, IL-6, IL-8, MCP-1, IFN-γ, VEGF, and IL-10 were found to be significantly higher in the severe and critical group than those in other groups. The plasma concentrations of IFN-α, IFN-γ, IP-10, MIG, and IL-6 were elevated in the severe and critical group at 5-10 days from symptom onset. Although the plasma concentrations of VEGF and IP-10 gradually decreased with time, their levels were significantly higher in the severe and critical groups throughout hospitalization.
In severe patients under the age of 60 do not show significant leukocyte alterations and express high IL-1RA, IL-6, CCL2, CXCL1, CXCL9, CXCL10, and EGF. In contrast, older patients express high CXCL8, IL-10, IL-15, IL-27, and TNF-α, presenting a significant reduction in the total T lymphocyte number and an increased expression of T cell exhaustion markers as compared to the younger [16,24-27].
Another study evaluated the cytokine profile in COVID-19 patients under oxidative stress, finding IL-6, IL-8, IL-10, VEGF, MCP-1 and EGF. This profile is compatible with critically ill patients, elderly or not, but with severity predictors such as T2DM, obesity, and insulin resistance [28].
In cases of Try starvation with critical shortage in tryptophan supply, the control of an immune response by IDO involves adjustable and versatile effects, including CD3ζ down-regulation and Treg generation by tryptophan deficiency and kynurenine production. CD4+CD25+ Treg phenotype through a process requiring GCN2 and leading to a gradual decrease in IL-2 production and up-regulation of IL-10 and TGF-β [8,29].
In agreement with what has been explained so far, it appears that the inflammatory storm in COVID-19 has an innate immunity profile that is highly mediated by monocytes and macrophages in patients who have severity predictors. Older people have more significant oxidative stress naturally related to the ageing process; 2TDM and obesity present polarization to M1 macrophages, tending to inflammation via IL-6 and Th17. In addition to underlying tissue hypoxemia, heart failure also has more tissue hypoxia. All these underlying pathologies shift metabolism to oxidative stress and perpetuate a chronic condition of inflammation with continuous production of IDO-1 and stimulation of the Tryptophan anti-inflammatory pathway with an increase in the Kynurenine/Tryptophan ratio leading to a state of tolerance due to KYN act on AhR receivers [13,30,31].
Furthermore, patients with insulin resistance have elevated serum 5-HT concentrations. Serotonin suppressed interferon (IFN-)γ-induced phagocytosis at high but had stimulatory effects at physiological IFN-γ concentrations. Serotonin suppressed the IFN-γ-induced antigen-presenting capacity of macrophage and suppressed the IFN-γ-induced MHC class II expression; Moreover, serotonin inhibited the production of TNF-α in LPS-stimulated peripheral blood mononuclear cells and increased the release of IL-1β. The upregulation of IL-1β, IL-6, and IL-8/CXCL8 secretion is 5-HT3 receptor-mediated. Activation of the 5-HT4 and 5-HT7 receptors increased the LPS-induced release of IL-1β, IL-6, IL-8/CXCL8, and IL-12p40, while on the contrary, it inhibited LPS-induced TNF- α release. 5-HT2B-mediated downregulation of T cell co-stimulatory [21,32-38] molecules and the simultaneous inhibition of IL-12 secretion. 5-HT2B activation results in modulation of monocyte-derived macrophage differentiation to acquire the anti-inflammatory M2 phenotype and inhibition of lymphocyte proliferation, tending to imbalance to tolerance with inhibition of TH17 and stimulus to Treg [39-42].
The pathophysiology hidden in COVID-19 is complex because, in addition to the infection influencing metabolic changes with an impact on the immune response, it is pathophysiology dependent on inflammatory comorbidities, intensified by the hypoxemia of underlying diseases and by the hypoxemia induced by lung lesions and in other tissues caused by SARS-COV-2 [43-45].
The thin, young patient without comorbidities may have a tryptophan deficit, but their shift to innate immunity should still be minor, ensuring adaptive immunity in action against the virus.
Obese patients and T2DM with insulin resistance trigger immunosuppression due to the inflammatory tendency of the underlying disease, underlying hypoxemia, hypoxemia stimulated by COVID-19 and by serum serotonin. There is an intense shift of metabolism to oxidative stress, intensified by the tryptophan deficit causing NAD/NADH+ depletion, compromising the machinery of aerobic respiration.
In the elderly, even without comorbidities, metabolism is already shifted to oxidative stress, as ageing is inflaming. In addition, adaptive immunity is also impaired with a decrease in T lymphocyte receptors due to immunosenescence.
It is also generally known that innate immunity has a more glycolytic profile, whereas adaptive immunity is highly dependent on aerobic respiration and ATP.
Thus, we have that the hypoxemia triggered by SARS-CoV-2 associated with Try depletion by internalization of intestinal ACE-2 are factors that lead to the intensification of the innate immune response, especially in individuals already predisposed to oxidative stress. This deviation causes intense immunosuppression, and it can also be magnified by the action of serotonin and the tryptophan deficit [12,46-48].
In yellow fever, deceased patients showed an increase in the Kynurenine/Tryptophan ratio. They showed a tendency to immunosuppression due to tissue hypoxemia. According to what is known about haemorrhagic shock in YF, it must have occurred due to DIC (disseminated intravascular coagulation), decreasing oxygen supply to tissues, producing adenosine, and shifting metabolism to oxidative stress [49,50].
In COVID-19, hypoxemia is already a fact given by the lung injury caused by SARS-CoV-2. Thus, all physiopathology of the host-parasite relationship is carried out amid hypoxemias. Consider that the more prolonged exposure to hypoxemia, the worse the immunosuppression, the tendency to innate immunity, and the oxidative stress pathway.
Thus, the signature of severity in COVID-19 is serotoninergic and oxidative [51-53]. Both signatures promote a shift from innate to adaptive immunity, mediated by monocytes with differentiation into dendritic cells and macrophages. Monocyte distribution, based on CD14+ CD16+ does not have significant change in the number of classic monocytes (CD14 ++ 16-ve), but an increase in intermediate (CD14++ 16+) and non-classic (CD14+ CD16++) subsets in patients compared to healthy controls. Shown classical double expression of positivity for HLA-DR/CCR2 and HLA-DR/CX3CR1 in patients with COVID-19; however, CCR2 expression was reduced in the intermediate subset in patients with COVID-19. Monocytes are heterogeneous and are classified into different subsets defined by the extent of their cell surface expression of CD14 and CD16. The main subset, termed classical monocytes, consists of CD14highCD16negative monocytes (CD14++ CD16−), while monocytes expressing CD16 are generally divided into an intermediate CD14highCD16low (CD14++ CD16+) subset and a CD14lowCD16high (CD14+ CD16+) subset not classic subset. Differential expression of the chemokine receptors CCR2 and CX3CR1 is associated with these human monocyte subsets with the classic CD14 ++ CD16− subset predominantly expressing CCR2 and the nonclassical CD14 + CD16 ++ subset showing lower CCR2 expression and significantly higher expression of CX3CR1. CCR2 and CX3CR1 are two chemokine receptors that regulate the responses of myeloid cells, such as monocytes and microglia, during inflammation. CCR2 and its ligand are crucial for the recruitment of inflammatory monocytes, increasing the adhesion of monocytes to the endothelium leading to their exit to the site of inflammation and polarizing macrophages into cells of the M2 repair tissue. The expression of CX3CR1 in intermediate and nonclassical monocyte subsets indicates a more phagocytic phenotype and a greater likelihood of endothelial damage. These cytokines appear to play an important role in CNS inflammation during viral encephalitis since inflammatory monocytes secrete proinflammatory cytokines such as IL-6, IL-1β and TNF-α, which have been implicated in the development of acute seizures and hippocampal damage [28,39,54-57].
In the gastrointestinal tract, mature gastrointestinal macrophages (GI-Mφ) maintain the homeostasis site through their hyperresponsiveness to the secretion of regulatory cytokines, including IL-10, which maintain regulatory site T cells (Tregs). In inflammatory bowel disease, the profile of these macrophages presents the CD11highCCR2+ CX3CR1+ phenotype. The expression of IL-10 mRNA in CD14+ CD16− (classic monocytes), but not CD14 + CD16 + (nonclassical monocytes) responds to lipopolysaccharide (LPS) stimulation, while TNF-α, IL-1 and IL transcripts -6 were detected in both subsets; Intermediate monocytes produce the highest level of IL-12 and IFN-γ in the context of antigen presentation [8].
Dendritic cells also play an important role in innate immunity in COVID-19 and can be classified as plasmacytoid DC (pDC), myeloid DC (mDC) and monocyte-derived DC (MDDC). pDCs directly block viral infections, resulting in the elimination of the pathogen, express TLR-7 and TLR-9 that detect viral and bacterial nucleic acids, and positively regulate the expression of MHC molecules and activation markers that will allow the antigenic presentation to T cells. PDC has been reported to activate CD8+ T cells and exhibit a low ability to activate CD4+ T cells. pCD differs in pCD1 and pCD2. pDC1 exhibits an immature phenotype with low to undetectable MHCII expression and activation markers, whereas pDC2 highly expresses MHCII and CD86. It has been reported that pDC1 induces T-reg, while pDC2 promotes proinflammatory T cell differentiation.
mDC is highly migratory and regularly travel from the periphery to lymphoid organs' T and B cell zones. At a steady-state or during infection, mDC regulates T cell functions. mDC are classified into two main populations based on the expression of CD141 (or blood DC antigen 3, BDCA-3) and CD1c (or BDCA-1 ): CD141+ DC (or cDC1) and CD1c+ DC (or cDC2). CD141 + DC have been shown to induce Th1, and CD8+ T cell differentiation, CD1c + DC has been found to generate not only Th1 but also Th2, Th17 and iTreg immune responses [58]. Mouse cDC2 cells have been reported as the primary inducers of the Th2 and Th17 immune response after antigenic stimulation. Mouse cDC2 produce IL-23 during infection, whether they are located in the lungs, skin or intestine.
DC-derived from CD16− and CD16+ monocytes are functionally distinct. CD16+ compared to CD16− MDDC expresses higher levels of CD86, CD11a and CD11c and higher levels.
Dendritic cells also play an important role in innate immunity in COVID-19 and can be classified as plasmacytoid DC (pDC), myeloid DC (mDC) and monocyte-derived DC (MDDC). pDCs directly block viral infections, resulting in the elimination of the pathogen, express TLR-7 and TLR-9 that detect viral and bacterial nucleic acids, and positively regulate the expression of MHC molecules and activation markers that will allow the antigenic presentation to T cells. PDC has been reported to activate CD8+ T cells and exhibit a low ability to activate CD4+ T cells. pCD differs in pCD1 and pCD2. pDC1 exhibits an immature phenotype with low to undetectable MHCII expression and activation markers, whereas pDC2 highly expresses MHCII and CD86. It has been reported that pDC1 induces T-reg, while pDC2 promotes proinflammatory T cell differentiation [40].
mDC is highly migratory and regularly travel from the periphery to lymphoid organs' T and B cell zones. At steady-state or during infection, mDC regulates T cell functions. mDC are classified into two main populations based on the expression of CD141 (or blood DC antigen 3, BDCA-3) and CD1c (or BDCA-1): CD141+ DC (or cDC1) and CD1c + DC (or cDC2). CD141 + DC have been shown to induce Th1, and CD8+ T cell differentiation, CD1c + DC has been found to generate not only Th1 but also Th2, Th17 and iTreg immune responses. Mouse cDC2 cells have been reported as the primary inducers of the Th2 and Th17 immune response after antigenic stimulation. Mouse cDC2 produce IL-23 during infection, whether they are in the lungs, skin, or intestine [21,59-61].
DC-derived from CD16− and CD16+ monocytes are functionally distinct. CD16+ compared to CD16− MDDC expresses higher levels of CD86, CD11a and CD11c and higher levels.
MDDC prepared with LPS expresses high levels of IL-12p40 mRNA and secretes high levels of IL-12. CD16-MDDC expresses high levels of the CD83 DC maturation marker after TLR2, TLR3 and TLR45 binding. CD1c - CD11c+ CD16+ CD14− cells that massively produce TNF-α, IL-12 and iNOS and highly express TLR7 and TLR8. DC produces high IL-23, IL-1β and IL-6, which lead to the development of Th1/Th17 cells. Because they are Th1/Th17 cell inducers, MDDC has been reported to have the ability to cross-present to naive T cells and transfer peptides from MHC-I to DC residing in lymphoid.
5-HT signalling on DCs. 5-HT acting through 5-HT1 and 5-HT2 receptors induced chemotaxis in immature human DCs and increased DC migration from lung to lymph node drainage in mice. 5-HT increased the production of the proinflammatory cytokine, IL-6, and the Th2 cytokine, IL-10, while it reduced the Th1 cytokine, IL-12p70. Furthermore, DCs treated with M5-HT increased their Th2 production by attracting the chemokine CCL22 while decreasing the chemokine Th1, CXCL10 mediated by 5-HT4 and 5-HT7 receptors. 5-HT treated CDCs induced a Th2 polarization in naive CD4 T cells. 5-HT modulated the differentiation of DCs from human monocytes, increasing the production of IL-10 but reduced the antigen-presenting capacity. The involvement of 5-HT2B shifts the TNF/CCL2 ratio to the anti-inflammatory side in LPS-treated macrophages [43,62-65].
The COVID-9 Th17 axis appears to be suppressed in severe cases. In patients with rheumatoid arthritis (RA) with dramatic disease showing an elevation of IL-6 and Th17, the levels of IL-1β and IL-18 directly correlate with IL-17. Inhibition of the IL-18/IL-18Rα signalling pathway inhibited the proliferation of autoreactive T cells and suppressed serum levels of IL-6, IL-18, TNF and IFN-γ. IL-18 is a cytokine of the IL-1 family proposed to promote barrier function in the intestine. IL-1 family cytokines are key co-regulators of CD4+ T cell fate, and the role of IL-1β in Th17 cell differentiation is mirrored by the contribution of IL-33 and IL-18 to Th2 and Th2 cell subsets. Although IL-18 is not essential for the differentiation of Th1 cells, in inflammatory conditions, IL-12 signalling promotes the expression of IL-18R1 in differentiating Th1 cells, after which IL-18 stimulation acts to increase the interferon production (IFN) -γ [66,67].
Based on an early review of the tryptophan listing and the articles on this metabolic flight, it offers yellow fever compared to what happens in COVID-19:
1- Critically ill patients evolve with a higher concentration of Kynurenine as metabolism is shifted to the inflammatory pathway.
2- In COVID-19, there is the internalization of intestinal ACE-2, with greater or lesser intensity depending on the amount in the inoculum, for example, or via (for example, upper respiratory tract or oral route), preventing the adequate uptake of tryptophan.
3- Lack of Try diverts metabolism to the kynurenine pathway and its metabolites, a pathway that is also influenced by vitamin B6.
4- Obese patients have serum serotonin due to insulin resistance.
5- Both Kynurenine and 5-HT have anti-inflammatory action and a tendency to immune response to Treg.
6- Hypoxemia and innate immunity to oxidative stress, as this pathway is preferentially glycolytic over adaptive, strongly dependent on aerobic respiration.
7- For this reason, we have two patient profiles, in general, in COVID-19: a) Insulin resistant or with some degree of previous hypoxemia or previous oxidative stress and b) Thin, young patients without comorbidities. Disease severity is related to the first group with a pre-existing inflammation profile.
8- Patients who express a tendency to the TH17 axis, with the production of IL-18, have a better evolution because they have a more inflammatory profile and are performed by adaptive immunity.
The depletion of tryptophan in COVID-19 patients due to the internalization of ACE-2 in the intestine promotes an unbalance of the immune response mediated by Kynurenine and magnified by 5-HT in patients with previous insulin resistance. The diversion of Tryptophan metabolism to the pathway of Kynurenine and its metabolites occurs due to Tryptophan shortage and hypoxia of the Disease and vitamin B6 deficiency, diverting metabolites for production of Kyn, HK, XA, and HaA. The action of Kynurenine, its metabolites, 5-HT, hypoxia, and the use of cathepsin L shift the metabolism to a tolerogenic environment, mediated by innate immunity, especially in critically ill patients. In Yellow Fever, there is no deficit in the absorption of tryptophan that triggers a more controlled cytokine storm, but still very lethal for those who develop tissue hypoxia resulting from DIC and shock. Those patients who present a profile tending to TH17 tend to have a better outcome, showing the importance of adaptive immunity in the viral and inflammatory control in the face of SARS-CoV-2 infection (Figure 4).
 Figure 4: The hypothesis of the representation of the relationship between tryptophan, Kynurenine and 5-HT in patients infected with yellow fever virus (red) and Sars-Cov-2 (green). (A) 5-HT x time curve; (B) Try curve x time. Figure A identifies that the group with insulin resistance has at baseline, higher serum 5-HT, which will lead to more significant immunosuppression and the mechanism of thermogenesis. B shows the Try curve that reaches lower serum concentrations due to impaired intestinal absorption due to the internalization of ACE-2 in COVID-19 patients. Lower concentrations of Try shift metabolism to form Kynurenine during inflammatory processes; (C) The figure shows Hypoxemia x disease severity. COVID-19 is a disease that causes intense hypoxia, taking the patient to gravity. In addition, when compared to yellow fever virus infection in COVID-19, hypoxemia starts from the earliest period of the disease. In contrast, in yellow fever, there is only more severe hypoxemia in patients who develop disseminated intravascular coagulation (DIC), causing tissue hypoxemia intensified by shock; (D) The Figure shows the hypothesis of serum Try concentration by ACE-2 internalization that is more intense when there is a viremia peak. Moreover, it shows viremia between severe and non-serious cases in COVID-19. Severe cases present more significant viremia and consequently greater internalization of ACE-2.
View Figure 4
Figure 4: The hypothesis of the representation of the relationship between tryptophan, Kynurenine and 5-HT in patients infected with yellow fever virus (red) and Sars-Cov-2 (green). (A) 5-HT x time curve; (B) Try curve x time. Figure A identifies that the group with insulin resistance has at baseline, higher serum 5-HT, which will lead to more significant immunosuppression and the mechanism of thermogenesis. B shows the Try curve that reaches lower serum concentrations due to impaired intestinal absorption due to the internalization of ACE-2 in COVID-19 patients. Lower concentrations of Try shift metabolism to form Kynurenine during inflammatory processes; (C) The figure shows Hypoxemia x disease severity. COVID-19 is a disease that causes intense hypoxia, taking the patient to gravity. In addition, when compared to yellow fever virus infection in COVID-19, hypoxemia starts from the earliest period of the disease. In contrast, in yellow fever, there is only more severe hypoxemia in patients who develop disseminated intravascular coagulation (DIC), causing tissue hypoxemia intensified by shock; (D) The Figure shows the hypothesis of serum Try concentration by ACE-2 internalization that is more intense when there is a viremia peak. Moreover, it shows viremia between severe and non-serious cases in COVID-19. Severe cases present more significant viremia and consequently greater internalization of ACE-2.
View Figure 4
This Praeludium, which introduces this possible pathophysiological pathway, introduces the theme of making an analogy with a FUGUE. The themes are also covered in the Appendix in a complementary way to help understand the theory.
For this review, articles in English and Russian were allowed (with translation from abstract to English). The PubMed and Google scholar databases were used, the second being for theoretical complementation when necessary. Thus, it was a database used throughout the manuscript's production. There was no characterization of the minimum date for the articles. From the descriptors: "Yellow Fever and Tryptophan"; "COVID-19 and Tryptophan"; "AhR and immune response"; "5-HT and immune response"; "5-HT and thermogenesis"; "Kynurenine and immune response"; "5-HT and monocytes"; "monocytes and macrophages"; "Obesity and Thermogenesis"; "COVID-19 and Cytokines"; "Th17 and Il-18"; "Treg and cytokines"; the first 20 articles were selected, the abstracts were evaluated so that the 5 best by theme were chosen. The criteria used were adapted from the PRISMA protocol, at https://www.canada.ca/en/public-health/services/reports-publications/canada-communicable-disease-report-ccdr/monthly-issue/2015-41 /ccdr-volume-41-04-april-2-2015/ccdr-volume-41-04-april-2-2015-3.html. Accessed in 02/09/2021. More 119 articles were add, during the writing, to embase the research.
The results are organised by topics regarding SARS-CoV-2 physiopathology and effects in the COVID-19 patient considering management possibilities. More information about it is in the Appendix and in the other articles published before [3,68,69].
Based on tryptophan as exposed above there are the necessity of reposition and use of drugs that maintain 5-HT at a good base line to perform its activities on CNS without psychiatric symptoms. There are 2 Clinical studies that use Fluvoxamine, a selective serotonin reuptake inhibitor (SSRIs) [70-72].
Regarding Phenylalanine it is important to considerer using drugs to supply catecholamine underproduction to maintain dopamine replacement and catecholamine intake. For patients with parkinsonian symptoms or psychiatric symptoms, try to replace dopamine orally (Levodopa) or intravenously in case of critically ill patients or in refractory shock. Maintain low doses of noradrenaline concurrently sustained by vasopressin [73].
Consider that cytokine storm causes generalized glandular hypofunction, just as it does in sepsis. Furthermore, as the patient's metabolism is shifted towards oxidative stress, cortisol and catecholamines, there is an important demand that the lack of Phe substrate cannot meet [2].
|
Recommendation based on clinical research (Try) e specialist opinion Prescribe: Citalopram 40 mg 1 time/day and Dopamine replacement. |
Permissive hypercapnia with a pH below 7.3 can be harmful to the patient by stimulating cathepsins and furins allowing greater viral replication and syncytia [74-77] formation in critically ill patients. Protective ventilation advocates 1) Tidal volume of 4-6 mL/kg of weight predicted by height; 2) Plateau pressure less than 30 cmH2O; 3) Driving pressure less than 15 cmH2O. 4) Tolerate higher CO2 targets with pH control (habitual conduct but prioritize less permissive values if possible) [78-81].
In relation to tolerating higher CO2 targets we must be careful not to allow low pH values. The adjustment of the pH must be done using the ventilator and, if necessary, use 8.4% sodium bicarbonate replacement to adjust the pH above 7.3. The expression of cathepsins at low pH also leads to immunosuppression by lymphocyte apoptosis. Diverting even more the microenvironment towards tolerance and permission to the development of neoplasms. More information on this topic can be found in an article recently submitted for publication "The Placental Buffer Effect and The Pathophysiology of COVID-19: Possibilities for a guide aimed at pregnant and postpartum women considering praxis: theory, clinical and laboratory observation".
|
Recommendations: Manage the ventilator to try to adequate PaCO2 values while prioritizing protective ventilation. Use 8.4% sodium bicarbonate to correct the pH so that it is always above 7.3. |
Replacement of vitamin B complex and N-acetylcysteine (NAC)Vitamin B complex replacement is necessary since COVID-19 evolves with Pellagra and changes in the B3 (niacin) production pathway are related to Tryptophan deficit with impaired aerobic respiration and shift to oxidative stress requiring replacement of pathways that are antioxidants. NAC does not perform well against some oxidant species such as H2O2, O2•−, OHNOO, and HO•, but for others, including NO2 and hypohalous acids, HOX, it could be more plausible. Hypochlorous acid (HOCl) and related species (hypobromous acid, HOBr; hypothiocyanous acid, HOSCN) are oxidants produced by activated neutrophils and monocytes through the activity of myeloperoxidase (MPO). Considering neutrophil chemotaxis due to the intense production of IDO-1, NAC replacement is necessary. NAC-derived cysteine is desulfurated to generate hydrogen sulfide. Also, NAC is necessary to the glutathione cycle. Furthermore, sulfane sulfur species produced by 3-mercaptopyruvate sulfurtransferase and sulfide. Some studies have been shown that quinone oxidoreductase are the current mediators of the immediate antioxidative and cytoprotective effects provided by NAC [58,82-85].
|
Recommendations: To prescribe N-acetylcysteine 600 mg/day Ascorbic acid 2 g/day If possible, prescribe Taurine. |
Neutrophils play an essential role in platelet activation, and, in COVID-19, severely ill patients evolve with marked leucocytosis due to neutrophilia. There are many reasons for Neutrophilia to happen, and most of it is stimulated by the expression of cathepsins, mainly cathepsin G.
Neutrophils enhanced the aggregation of human platelets in vitro in a dose-dependent fashion, and this effect was diminished by pharmacologic inhibition of cathepsin G activity and knockdown of cathepsin G expression (Figure 5).
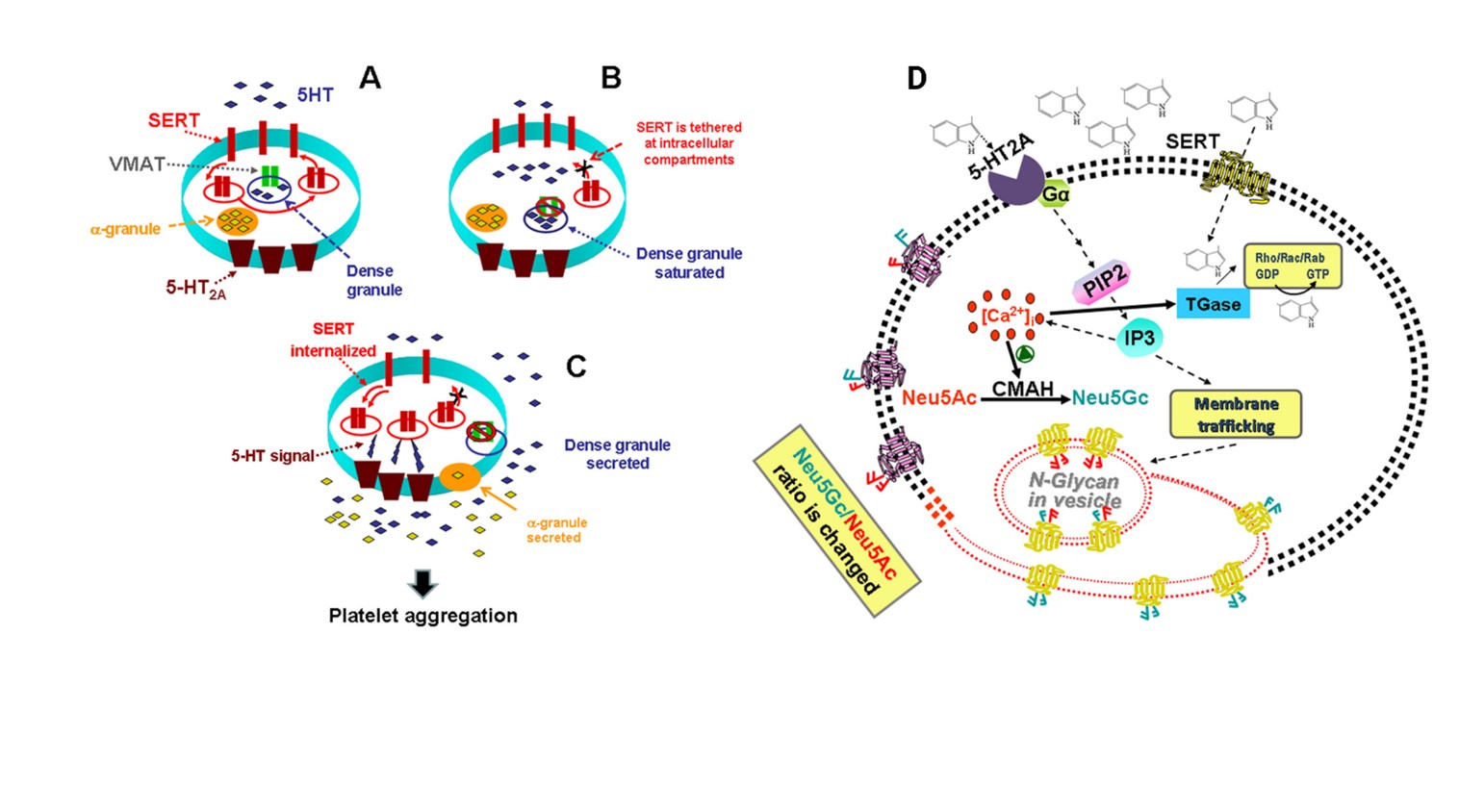 Figure 5: (A) High 5-HT leads to abnormalities in platelet trafficking of SERT, which reduces the density of SERT molecules in the plasma membrane; (B) Based on our findings for the known actions of 5-HT on platelets, we propose that high levels of uptake lead to saturation of dense granules and 5-HT appears in the cytoplasm of platelets. VMAT is disabled and can no longer remove 5-HT in dense granules. This leads to serotonylation of small GTPases such as Rab4, Rho and Rac via TGase activated by Ca2+. In the form of GTP, Rab4 binds and prevents translocation of SERT to the plasma membrane; (C) These processes are involved in platelet activation and aggregation in a two-step process: in the first step, elevated 5-HT controls platelet 5-HT uptake rates by altering SERT plasma membrane trafficking which, in turn, raises plasma Level 5-HT further; then, in the second step, the elevated plasma level of 5-HT activates 5-HT2A, which accelerates the exocytosis of α granules. Secretion of prothrombotic molecules from α granules into plasma with high levels of 5-HT propagates thrombus formation. However, even at the highest levels of plasma 5-HT, there are always several SERT molecules in the plasma membrane that continue to clear plasma 5-HT, but at a lower rate, probably until the plasma 5-HT level returns. at the physiological level. (D)The stimulation of cells with 5-HT activates, via 5-HT2A signalling, the production of Neu5Gc via the catalytic function of CMAH. 5-HT signalling is mediated by the G protein-coupled 5-HT2A which facilitates the formation of Inositol 1,4,5-triphosphate (IP3) resulting in a rise of cytoplasmic Ca2+ in platelets. Intracellular elevated Ca2+ activates CMAH, which elevates the number of Neu5Gc containing N-glycans on the plasma membrane of platelets. All the figure showed that 5-HT signalling is important either on trafficking of Neu5Gc (presented in the diagram in blue) containing vesicles to the membrane plasma or enhancing the catalytic ability of CMAH or both in inducing platelet activation and aggregation.
View Figure 5
Figure 5: (A) High 5-HT leads to abnormalities in platelet trafficking of SERT, which reduces the density of SERT molecules in the plasma membrane; (B) Based on our findings for the known actions of 5-HT on platelets, we propose that high levels of uptake lead to saturation of dense granules and 5-HT appears in the cytoplasm of platelets. VMAT is disabled and can no longer remove 5-HT in dense granules. This leads to serotonylation of small GTPases such as Rab4, Rho and Rac via TGase activated by Ca2+. In the form of GTP, Rab4 binds and prevents translocation of SERT to the plasma membrane; (C) These processes are involved in platelet activation and aggregation in a two-step process: in the first step, elevated 5-HT controls platelet 5-HT uptake rates by altering SERT plasma membrane trafficking which, in turn, raises plasma Level 5-HT further; then, in the second step, the elevated plasma level of 5-HT activates 5-HT2A, which accelerates the exocytosis of α granules. Secretion of prothrombotic molecules from α granules into plasma with high levels of 5-HT propagates thrombus formation. However, even at the highest levels of plasma 5-HT, there are always several SERT molecules in the plasma membrane that continue to clear plasma 5-HT, but at a lower rate, probably until the plasma 5-HT level returns. at the physiological level. (D)The stimulation of cells with 5-HT activates, via 5-HT2A signalling, the production of Neu5Gc via the catalytic function of CMAH. 5-HT signalling is mediated by the G protein-coupled 5-HT2A which facilitates the formation of Inositol 1,4,5-triphosphate (IP3) resulting in a rise of cytoplasmic Ca2+ in platelets. Intracellular elevated Ca2+ activates CMAH, which elevates the number of Neu5Gc containing N-glycans on the plasma membrane of platelets. All the figure showed that 5-HT signalling is important either on trafficking of Neu5Gc (presented in the diagram in blue) containing vesicles to the membrane plasma or enhancing the catalytic ability of CMAH or both in inducing platelet activation and aggregation.
View Figure 5
Cathepsin G increased platelet surface expression of P-selectin (an activation-dependent neutrophil binding site), the glycoprotein IIb/IIIa complex (fibrinogen receptor), and glycoprotein IV (thrombospondin receptor), and decreased surface expression of glycoprotein Ib (von Willebrand factor) (Figure 5) [86-88]. Serotonin has a mitogenic effect on megakaryocytopoiesis. This effect may be mediated via the 5-HT2 receptor, which is known to be coupled to G protein. Platelet α-granule constituents, including platelet-derived growth factor (PDGF), platelet factor 4 (PF4) and transforming growth factor-β (TGF-β), can affect megakaryocytopoiesis. Serotonin, a platelet dense granule constituent, has been shown to have a mitogenic effect on fibroblasts and smooth muscle cells, but whether it has the same effect. Circulating metastatic cells attract platelets and influence them to release their granule content acting in human NK cells losing their cytotoxicity and their ability to produce IFN-γ (interferon-γ), possibly through the downregulation of NK G2D ligand-mediated by platelet-TGF-β (transforming growth factor-β) release. Platelets' immune-modulatory potential is dependent on the underlying pathological conditions, as platelets from patients with dengue virus infection stimulate monocytes to produce MCP-1, IL-1β, IL-8 and IL-10 [89,90].
Myeloperoxidase (MPO) is the most abundant protein in neutrophils and represents 5% of their total protein content. MPO and neutrophil activation via CD11b/CD18 integrins, which is an indicator of MPO's possible contribution to neutrophil recruitment to the site of inflammation. A study shows that SARS-CoV-2, which we detected significantly higher expressions of MPO (~4 fold, p < 0.05) at the nasopharynx region, and speculates this neutrophil burst and even more overexpressed MPO may cause the production of excess hypochlorous acid (HOCl) and other reactive oxidants that also damage the nasopharynx tissue. Only MPO may be an important factor where the protective inflammation can become pathological in SARS-CoV-2 cases.
In previous work not yet published, I exposed the critical role of Adenosine in the pathophysiology of COVID-19. Adenosine is produced to antagonize mechanisms of cellular aggression, in the case of COVID-19, related to intense hypoxia caused by the viral attack, triggering lung lesions and other target tissues [91-93].
The most critical issue related to Adenosine is the production of ADP (adenosine diphosphate), which is a platelet activator and stimulates inflammation. Moments of support for this metabolic pathway, such as a low oxygen supply, can be stimuli for the maintenance of inflammation in the patient COVID-19. This fact is seen in clinical practice when lowering the O2 fraction in patients, as they restart an inflammatory process with leucocytosis, increased LDH, D-dimer, for example.
The adenosine A1 receptor, which has a relatively high affinity for Adenosine, promotes neutrophil chemotaxis, and the A2A and A3 receptors are expressed at high levels on neutrophils, where they suppress neutrophil effector functions such as reactive oxygen species (ROS) formation when activated [94-98].
Dipyridamole inhibits NETosis and prevents thrombosis, in addition to acting to reduce inflammation. I have no experience with this drug, but I think a clinical study is needed as soon as possible.
|
Recommendations based on the author's literature and experience
a) For ICU patients Use of heparin pump in. Acetylsalicylic acid 100 mg once daily. Clopidogrel 75 mg once daily.
b) For inpatients Prophylactic heparin or enoxaparin. Acetylsalicylic acid 100 mg once daily. Clopidogrel 75 mg once daily
c) For clinically symptomatic patients without the need for hospitalization: Assess risk factors (acute myocardial infarction, previous stroke, previous thrombosis) for monotherapy or dual therapy - suggested time: 1 month after the end of the acute disease (about 14 days after the onset of symptoms).
Prophylactic heparin or enoxaparin. Acetylsalicylic acid 100 mg once daily.
All with Citalopram 40 mg, once a day for 30 days. |
Initially, it was thought that anaemia in COVID-19 was due to hemophagocytic syndrome or Histiophagocytic Syndrome, but few cases present these associated diseases. This fact does not make any of the previous works unfeasible, as we are talking about science and something new to us, never seen before. Today it is one way; tomorrow, everything can change. The hypotheses must be launched and, it was from these studies, we had many important parameters to be seen in the laboratory tests of COVID-19 patients [14,99]. Iron is a cofactor of tyrosine hydroxylase and tryptophan hydroxylase, enzymes responsible for synthesising dopamine and serotonin, respectively. IFN-γ, one of the central cytokines of Th1 type immune response, activates IDO and neopterin formation in hematopoietic stem cells and influences the proliferation of various stem cell populations. The intravenous injection of neopterin into mice results in a prolonged decrease in the number of erythroid progenitor cells and an increased number of myeloid progenitor cells (CFU-GMs) by activating stromal cells. QUIN inhibits EPO production by stimulating the production of nitric oxide (NO) and inducing HIF-1α degradation. Trp metabolites like Kyn, on the other hand, increase hepcidin expression and inhibit erythropoietin (EPO) production by activating AhR. AhR competes with hypoxia-inducible factor 2α (HIF-2α), the key regulator of EPO production, for binding with HIF-1β. In the liver, serotonin represses hepcidin is through a 5HT2B receptor-dependent pathway, independently of any other known hepcidin regulators, including bone marrow signals. This regulation is conserved in humans and shows physiological significance as a negative correlation between serotonin and hepcidin levels [14,100].
In COVID-19, persistent anaemia seems to be related to:
1- Most seen in obese and insulin-resistant patients. Modification of Tryptophan metabolism in inflammation that is carried out by the IDO-1 enzyme. In SARS-CoV-2 infection, the production of IDO-1 is produced in large quantities and, for its formation, pyrrolic rings are needed, becoming in deficit for the construction of new erythrocyte molecules.
IDO1 is composed of two domains, among which the function of the N-terminus domain is still unclear and may help the stability of the system, while the large C-terminus domain contains the active heme centre performing the core functions of the enzyme. The C-terminus domain is rich in hydrophobic residues, which shows a component strictly to the shape of the indole ring of the substrate, allowing the interaction of oxygen molecule (O2) with the iron atom (Fe) in the first step of the reaction. In the large domain, there is a ligand delivery tunnel for O2 and water molecules (H2O), which extends along with the E and Fα helix to the active heme centre, as well as the 360-381 loop region controlling the shuttle of substrate/product in/out the catalytic site. The loop parallels with the heme plane before adding inhibitor but moves to the small domain (i.e., N-terminus domain) after the association with IDO1/Try/kynurenine. The structural biology of IDO1 will be detailed described in the section [101,102].
Reinfusion of autologous hematopoietic peripheral blood stem cells (PBSC) or bone marrow is often accompanied by flushing, dyspnoea, abdominal cramping, nausea, and diarrhoea. These symptoms and the observation that they can be prevented by ondansetron, a selective 5-HT3 receptor antagonist, led to the assumption that these side effects are due to the infusion of free serotonin during the reinfusion of PBSC or bone marrow.
2- More seen in thin patients without insulin resistance. Tph1 as an erythroid gene: moreover, it uncovered a fundamental role for 5-HT in regulating the hematopoietic stem cell fate along the erythroid pathway. EPO induces TPH1 expression and 5-HT synthesis necessary for erythroid progenitors survival and proliferation. In purified human CD36+ cord blood cells, using qRT-PCR, we demonstrate that mRNA for TPH1, the 5-HT2A receptor (5-HT2AR-HTR2a), and the 5-HT-specific membrane transporter (SERT-slc6a4) were highly expressed at the specific pro-erythroblast stage of differentiation (day 3 of the culture after CD36+ isolation). These patients seem to develop some myelodysplasia; for this reason, it is vital to perform a therapeutic test with Citalopram and replace Tryptophane and, logically, to follow these patients properly, since the fact that SARS-COV-2 increases the expression of cathepsins is possible that neoplastic diseases are more likely to develop, especially considering a tolerant environment.
IFN-γ, one of the main cytokines of Th1 type immune response, activates IDO and neopterin formation in hematopoietic stem cells and exerts an influence on the proliferation of various stem cell populations. Trp metabolites like Kyn, on the other hand, increase hepcidin expression and inhibit erythropoietin (EPO) production by activating AhR. AhR competes with hypoxia-inducible factor 2α (HIF-2α), the key regulator of EPO production, for binding with HIF-1β. Well in line with this finding, Kyn/Trp and neopterin were shown earlier to be associated inversely with haemoglobin concentrations and positively with hepcidin concentrations in patients with HIV-infection before antiretroviral therapy. Antiretroviral treatment slowed down immune-mediated Trp catabolism and improved iron metabolism and anaemia [14].
It is necessary to be careful and attentive when administering Citalopram to type 2 diabetic patients with insulin resistance, as they may have symptoms like those seen in reinfusion syndrome, which occurs in autologous bone marrow transplants. In outpatient follow-up, I had the opportunity to witness the complaint of diarrhoea and abdominal pain after a week of the introduction of Citalopram.
I advise not to prescribe Citalopram for these patients, but it is essential to replace Tryptophane and melatonin. Usually, with these medications, the patient presents an improvement of symptoms related to the lack of intestinal absorption of the amino acid in question. It is important to ensure that these patients are not under the action of inflammation so that Try metabolism is performed via hepatic TDO.
|
Recommendations:
Folic acid 5 mg/day
Block inflammation with corticosteroids: I recommend a pulse between 250 mg and 1 gram of Methylprednisolone and reassess the need for a new pulse and, if necessary, with lower doses, as the immunosuppression profile of patients is very heterogeneous*.
Block the inflammation triggered by the hypoxia pathway, allowing the patient to be under greater offers of FioO2.
Prescribe Citalopram 40 mg once a day. Observe symptoms of excess serotonin described below.
*In chronic patients: Urine and Blood cultures are necessary. Sometimes BAL is required too. |
The General and Integrated pathophysiology: SARS-CoV-2 marked the year 2020 because it caused the pandemic that is still ongoing in 2021. To date, we do not have any specific antiviral drugs, nor even other antiviral medicines that are effective. So far, we have only used corticosteroids to change the outcome of inflammation when used promptly, selective serotonin inhibitors and perhaps vitamin B3 supplementation, the anti-aggregating action of acetylsalicylic acid and anticoagulants such as heparin.
The disease is difficult to manage, and the explanation for this is that SARS-CoV-2 modifies some metabolic functions that imply the sum of multiple factors that support inflammation. Inflammation alters the microenvironment by shifting the balance of some reactions to the formation of undesirable by-products that are toxic to many tissues. COVID-19 is disease-specific to each person and, at the same time, bears similarities between the different patient profiles that we observe. We have a disease, the severity of which is related to inflammatory comorbidities, ageing and tryptophan reserves. After all, from the new evidence it is possible COVID-19 is a viral disease that evolves to a Tryptophan (Try) syndrome.
In general, SARS-CoV-2 infects any tissue that expresses the angiotensin-2 converting enzyme protein (ACE-2) on the surface of its cells. This enzyme is responsible for the balance of inflammation linked to angiotensin since the internalisation of ACE-2 provides greater availability of angiotensin II, which presents itself as an inflammatory agent by stimulating AT1 receptors (inflammatory) in cells of innate and adaptive immunity, adaptive in addition to favouring increased vascular resistance and increased aldosterone. However, there is an anti-inflammatory effect when it binds to the AT-2 receptor. Thus, we have an ACE/Ang-II/AT-1 results in increased pro-inflammatory cytokines, including IL-1, IL-6 and TNF-α, intensified by the activation of innate and adaptive immune and ACE2/Ang- (1-7)/MasR axis down-regulation in the elderly, hypertensive, diabetic and cardiovascular compromised patient [1-6].
SARS-CoV-2 produces damage to the organs it passes through, and related injury presents generalized vascular endothelium injury, lung injury with airways inflammation and alveolar zones with hyaline membranes and type 2 pneumocyte hyperplasia, interstitial fibroblastic proliferation with fibromyxoid stroma, organising pneumonia, squamous metaplasia, and alveolar wall acute inflammation, diffuse hyaline membranes. This injury leads to hypoxemia. The internalization of the intestine ACE-2 stops capturing tryptophan, and the supply of this amino acid is now deficient for the formation of NAD/NADH+ (niacin) [20,21]. At the same time, interferon (IFN)-γ in dendritic cells (DC), macrophages, and monocytes due to IDO-1, while in a medium in homeostasis and without inflammation, metabolism tryptophan is carried out by the liver from TDO-1. This change provides for the diversion of the metabolic cascade towards Kynurenine production. Tryptophan metabolism via the KP results in a neurotoxin, quinolinic acid (QA), and a neuroprotective compound, kynurenic acid (KA). KA binds to the glutamate recognition site of the N-methyl-D-aspartate (NMDA) receptor and antagonizes it, while QA binds to the glycine site of the NMDA receptor with agonistic properties [22-25].
The most common and least severe side effects of niacin deficiency include depression, apathy, anxiety, headache, fatigue, disorientation, and memory loss. Severe niacin deficiency can cause a potentially fatal disease called pellagra. If left untreated, it can cause skin conditions, diarrhoea, dementia, and death. Niacin is part of many metabolic reactions in the body, so its depletion is related to the systemic symptoms present in COVID-19. Some studies report symptoms related to atherothrombotic mechanisms, as niacin can improve endothelial function, vascular inflammation, and vascular regeneration. Independent of correcting dyslipidaemia, niacin improves endothelial tube formation under lipotoxic and hypoxic conditions. Niacin improves HMVEC angiogenic function under lipotoxic and hypoxic conditions [8,11-13].
Vitamin B3 can be formed by ingesting NA and Nam and by the tryptophan route. The intracellular pathway is reduced to NADH+, coexisting between the forms Nicotinamide adenine dinucleotide (NAD) and NADH+, or phosphorylated in NADHP, which is recycled or excreted. There are three ways to obtain NAD: Preiss - Handler pathway (PHP): NAD of nicotinic acid; via "de novo": tryptophan NAD and rescue route: Nam's NAD (Figure 1). NAD is an essential cofactor involved in many body reactions due to its reducing potential and participating in several energy metabolism pathways, including glycolysis, β-oxidation, and oxidative phosphorylation. NAD is a necessary cofactor for post-translation modifications, such as ADP-ribosylation and deacetylation by poly(ADP-ribose) polymerases (PARPs) and Sirtuins. NAD regulates energy metabolism, DNA damage repair, gene expression and stress response through enzymes. NAD(+) is the oxidised form of the nicotinamide adenine dinucleotide found in all living cells. In metabolism, NAD(+) is involved in redox reactions. NAD+ is degraded in the ADP-ribosyl transfer reactions as part of other noteworthy reactions. The lack of NAD is responsible for increasing neuronal degradation and the expansion of its precursor, nicotinamide mononucleotide (NMN), related to neuronal toxicity.
Another extra pathway in mammals can form NAD is the Sirtuins pathway, a family of mammalian NAD + dependent lysine histone deacetylases that are members of the highly conserved class III histone deacetylases and seven Sirtuingenes (Sirtuins 1-7). This pathway does not use Zn2+ but uses NAD and nicotinamide adenine dinucleotide as cofactors in a family of mammalian NAD+ -dependent protein lysine histone deacetylases (SIRT3, SIRT4 and SIRT5) [21,30-42].
Some studies also show the role of nicotinamide (NAM) as an inducer of autophagy in cells of tissues injured by hypoxia by inhibiting the mammalian target of rapamycin (mTOR), so the cells can remain viable, avoiding apoptosis. mTOR pathway is a downstream target of the adenosine monophosphate-activated protein kinase (AMPK), and the regulation on this pathway exerts a substantial effect on cell viability. NAM can modulate cellular energy metabolism by regulating AMPK.
In the first days of infection, the inflammatory process initially inhibited by viral escape mechanisms starts to have its function normalised with the end of viremia because even if it escapes by inhibiting IFN-gamma and TNF, this process does not last more than 72 hours. There are cytotoxic lymphocytes that act against the virus and local defence cells, such as macrophages, dendritic cells, and monocytes. Therefore, damage to the vascular, pulmonary endothelium and other infected tissues already exists and causes respiratory and systemic consequences [3].
Structural changes in the organs infected by SARS-CoV-2 occur about D14 after infection or (D7 after the onset of the first symptoms). After this phase, the patient is symptomatic for 2 to 3 days without noticing hypoxemia or maintained hypoxemia with rare worsening. After this period, two paths are followed: an intense aggravation mediated by a neutrophil march in which the patient has a severe illness and often his condition is critical. Or the disease presents as moderate or oligosymptomatic forms. The release of CXCL8 (human homologue of murine chemokine CXCL2) by mononuclear cells, Sirtuin (SIRT6), and 5-HT are responsible for the neutrophil attraction.
Exposure to hypoxemia determines the consumption of NAD/NADH+, and its decrease in intracellular concentrations is responsible for the activation of the inflammatory response or even its exacerbation. The lack of niacin (NAD/NADH+) compromises aerobic respiration, while inflammation promotes the production of IDO-1 by phagocytes and platelet activation for the reconstruction of injured vessels tissues. Platelet activation is responsible for the release of serotonin, as it is one of the main reservoirs of this molecule in the human body. Hypoxia is an essential factor in the microenvironment commonly seen in many pathological contexts. DCs that reside in tissues and precursors of blood DC, including monocytes, can be recruited to sites of inflammation or solid tumour tissues that may be hypoxic. DCs in solid tumours are tolerant and in the joints of patients with RA inhibit adaptive Th1 cells while stimulating inflammatory Th17 cells. COVID-19 represents the struggle between tolerance and inflammation.
Tolerogenic DCs perform immunosuppressive functions through inhibitory molecules, such as programmed death-ligand receptors (PD-L1 and PD-L2), CD103, arginase, 2,3-dioxygenase indoleamine (IDO). Hypoxemia is fundamental to the patient presents immunosuppression due to COVID-19. The immunosuppression magnitude depends on the patient's time of exposure to hypoxemia. For this reason, "intubation in time" is part of the treatment to be offered to a profile of patient. Dendritic cells (DCs) can differentiate from monocytes via GM-CSF, IL-4 takes approximately seven days under normoxic conditions (21% O2), and IDO-1 expression is upregulated in a hypoxemic environment. Hypoxemic tissue produces adenosine, an intermediate metabolite of nucleic acids. Adenosine is a key signalling molecule that orchestrates cellular response to hypoxia, energy depletion, and tissue damage by activating its G protein-coupled receptors (GPCR) on multiple cell types. The gamma interferon production stimulates the monocytes to produce IDO-1 and migrate to the injured tissue. They find adhesion factors and colony-stimulating factor (CSF-1), granulocyte-macrophage colony-stimulating factor, IL-3 and others. In tissues, monocytes are differentiated into macrophages and dendritic cells, which, together with the cells that reside in the tissue, play an essential role in eliminating pathogens. Circulating monocytes will differentiate into tissue macrophages that can be broadly classified into two groups: M1 macrophages after activation by IFN-γ or lipopolysaccharides (LPS) or M2 activated macrophages [103-106].
M2 macrophages subdivide into M2a IL-4 or IL-13), M2b (immune complexes in combination with IL-1β or LPS) and M2c (IL-10, transforming growth factor β or glucocorticoids). Activation of macrophages with IFN-γ, TNF, and IL-1β positively regulates the first rate-limiting enzyme of the kynurenine (KP) pathway, indoleamine 2,3-dioxygenase (IDO).
Maintenance of aerobic respiration is dependent on NAD/NADH+ dependent on exogenous niacin (Nam or NA) for their biosynthesis, and its concentration is inversely related to the production of TNF-alpha. For this reason, vitamin B3 has anti-inflammatory characteristics, as it acts in the replacement of NAD/NADH+. SIRT6 also plays an essential role in stimulating TNF-alpha expression [19,47,107-109].
NAD+ is produced by the salvage pathway during hypoxemia and is metabolised by ectoenzymes to form adenosine (ADA) that regulates the immune response, promoting vasodilatory and antivascular endothelial leakage by increasing IDO-1 released by DCs. Hypoxia-inducible factor-1 (HIF-1 increases T-cell activation and alters cell metabolism favouring their differentiation to effector cells. IDO-1 inhibits cell proliferation and increases the p53 transcriptional targets p21 and TP53-induced glycolysis and apoptosis regulator. IDO decreases the transcriptional targets of both HIF-1α and c-Myc, hexokinase II and lactate dehydrogenase-A.
It is necessary to consider that the COVID-19 severity occurs in people with comorbidities of an inflammatory profile but the greater intensity in obese, diabetic, and older adults. The first two are inflamed by adipocytes' characteristics producing inflammatory cytokines and in the elderly by immunosenescence with the natural increase in oxygen reactive and progressive loss of cellular repair mechanisms [110-113].
Serotonin (5-hydroxytryptamine, 5-HT) is a monoamine that mediates a range of central and peripheral functions. Synthesis of serotonin in both locations relies on the enzyme tryptophan hydroxylase encoded by two different genes, Tryptophan hydroxylase 1 (Tph1) and Tryptophan hydroxylase 2 (Tph2) expressed in the periphery and in the brain, respectively. Serotonin does not cross the blood-brain barrier, and thus each pool of this molecule has its distinct functions. In adipocytes, intestinal-derived serotonin (GDS) signals through the Htr2b receptor to favour lipolysis, increasing phosphorylation and hormone-sensitive lipase activity. In hepatocytes, GDS signalling through Htr2b promotes gluconeogenesis by increasing the activity of two rate-limiting gluconeogenic enzymes, FBPase and G6Pase. Also, GDS signalling in hepatocytes prevents glucose uptake in a manner dependent on Glut2, further favouring the maintenance of blood glucose levels.
Ang II induces its pleiotropic vascular effects through NADPH-driven generation of reactive oxygen species (ROS) that increase intracellular free Ca2+ concentration ([Ca2+]i), a major determinant of vascular reactivity. The key mechanism is the generation of ROS by serotonin degradation, catalysed by the mitochondrial enzyme MAO-A. Oxidative stress can act as a second hit in the pathogenesis, whereas lipid accumulation represents the first hit. In mitochondria, ROS induce damage to mitochondrial DNA30 and enzymes of the respiratory chain. Furthermore, they are potent inducers of the mitochondrial transition pore, leading to ultrastructural changes such as mitochondrial swelling, the formation of megamitochondria, cytochrome c release, and cell death. Lipid peroxides act as strong chemoattractant for neutrophils that are responsible for NETs and an intense ROS mechanism [114-116].
The oxidative stress could be a critical molecular linkage between the hypothalamic-pituitary-adrenal (HPA) axis dysfunction and mental disorders. The stress-induced increase in cortisol levels accelerates glucose metabolism and the production of reactive oxygen species. During stress, the hypothalamic-pituitary-adrenal HPA axis becomes critically engaged through its role in activating glucocorticoids' release, with consequent increases in heart rate, blood pressure, and metabolism. Acute release of cortisol during stress is responsible for enhancing cardiovascular function, mobilisation of energy, inhibition of growth and reproductive functions, and some immunological responses. However, glucocorticoid secretion's adaptive advantages during stress are limited to its acute rather than chronic release. Chronic elevation of cortisol is harmful, resulting in insulin resistance, visceral fat deposition, osteopenia and osteoporosis, inhibition of T helper-1 directed cellular immunity, and chronic suppression of the mesolimbic dopaminergic reward system.
COVID-19 evolves severely in this patient profile. The accumulation of fatty tissue associated with chronic stress with insulin resistance provides an explosive environment that, at the expense of hypoxemia, consumes NAD/NADH+ shifting the balance to an inflammatory process and even more dependent on beta-oxidation of fats. In these patients the large amount of peripheral serotonin is initially used for ROS production, which, when degraded, allows stored fat to be consumed in a thermogenic process that causes tissue damage and hyperthermia. Serotonin consumption leads to its lack of due to internalisation of ACE-2 and less tryptophan available.
Serotonin depends on reserves, so mast cells and platelet activation try to supply the physiological need for this molecule. A serotonin discharge may occur in each new platelet activation and symptoms of agitation, diarrhoea, and increased body temperature-Serotonin syndrome [32,117,118].
Vitamin B3 reserves are also depleted, and due to hypoxemia, MPO is inactivated, and Kynurenine has its products converted into NAD. However, the hypoxemic medium is the food for maintaining the beta-oxidation process that attracts more neutrophils -also chemo- attracted by activated monocytes. With each new tissue injury, a new activation of platelets and thus the inflammatory process is maintained. There are several neutrophil attraction mechanisms, but the maintenance of beta-oxidation associated with monocyte activation seems to play a dominant role since serotonin is depleted. However, in insulin-resistant patients 5-HT shows a significant role in the neutrophil marching [37,119-121].
Thermogenesis (TG) is frequent in patients with insulin resistance; they are severe and critical, usually in ICUs. These patients remain at elevated temperatures even with the use of antipyretic medications. TG occurs and causes a high temperature in the patients, not in fever but hyperthermia. These patients have excess serum serotonin, but it is quickly consumed and poorly restored due to the consumption of Tryptophan for the Kynurenine pathway, consumption of BH4 and Try deficit due to the internalization of ACE-2.
bα1AR, α1-adrenoceptor; β3AR, β3-adrenoceptor; BAT: Brown Adipose Tissue; Ca2C: Calcium Ion; FADH2: Flavin Adenine Dinucleotide; MAOI: Monoamine Oxidase Inhibitors; LSD: Lysergic Acid Diethylamide; MDMA: Ecstasy, Methylenedioxyamphetamine; NADH: Nicotinamide Adenine Dinucleotide; NST: Non-Shivering Thermogenesis; NE: Norepinephrine; RYR1: Ryanodine Receptor; SSRIs: Selective Serotonin Reuptake Inhibitors; SERCA: Sarcoplasmic/Endoplasmic Reticulum-Atpase Pump; SKM: Skeletal Muscle; SNS: Sympathetic Nervous System; UCP1: Uncoupling Protein 1; UCP3: Uncoupling Protein 3; TCA: Tricyclic Antidepressant.Insulin regulates plasma glucose levels by suppressing gluconeogenesis in the liver and inducing glucose uptake in skeletal muscle and adipose tissue. Obesity causes insulin resistance and glucose intolerance. The βcells overproduce insulin and augment their mass to compensate for insulin dysfunction. When it fail eventually results in type 2 diabetes. Plasma glycerol, produced by adipose tissue and used for gluconeogenesis, is not increased in gut-specific Tph1 during food deprivation [37,105,122,123].
5-HT increases hepatic gluconeogenesis; it simultaneously decreases glycogen synthesis and reduces GLUT2-mediated uptake of glucose. Exogenous 5-HT attenuates the thermogenic potential of the β-adrenergic receptor agonist, isoproterenol. 5-HT reduces cAMP levels in iBAT, lowers HSL activation, and reduces the expression of uncoupling protein 1 (UCP1), the mitochondrial protein responsible for thermogenesis). Peripheral 5-HT augments obesity by suppressing the "browning" of white fat, reducing UCP1 in BAT, and suppressing the secretion and expression of metabolically beneficial adiponectin [124-126].
Different mesenchymal progenitors form WAT and BAT, with BAT cells coming from progenitors common to the skeletal muscle tissue, which are favourable for myogenic factor 5 (Myf5), whereas WAT cells come from progenitors lacking this factor. A decrease in BAT activity is observed in individuals with obesity and type 2 diabetes, which is thought to exacerbate weight gain and metabolic disease by lowering energy expenditure [127-130].
Norepinephrine (NE) binds to β-3-adrenergic receptors (β3-AR) in adipocytes to activate thermogenesis. Cold causes the activation of specific channels located in thermoreceptor neurons innervating the body's, signalling for the release of NE. NE binds β3-AR activating adenylate cyclase, activating cAMP-dependent protein kinase A, promoting lipolysis and mitochondrial biogenesis by expressing several genes, including lipases, AMPK UCP1 and PGC1α [124,131,132].
Cold exposure increases the up-regulation and translocation of glucose transporters, glucose transporter (GLUT) 1 and 4, facilitating glucose uptake from plasma. This axis protects against hypertriglyceridemia, hyperglycemia and insulin resistance, which are common disorders associated with obesity. Additionally, cold exposure increases the production of vascular endothelial growth factor (VEGF), which enhances angiogenesis and provides a venue for heat dissipation.
There are a balance between leptin production and thermogenesis. Leptin may increase lipolysis and browning, although much less than upon SNS activation, and many of the hypothalamic neurons involved in the regulation of thermogenesis are also leptin sensitive. It appears that leptin has a double role in promoting browning and lipolysis, but after cold exposure, the decrease in leptin expression is compensated by other thermogenic drivers indicated before [124,133,134].
BAT is acutely activated by cold exposure via stimulation of the sympathetic nervous system (SNS), increasing BAT's intracellular cyclic adenosine monophosphate (cAMP) level. The high level of cAMP then increases protein kinase A (PKA)-mediated lipolysis of intracellular TAG into free fatty acids (FFA) to provide fuel for β-oxidation. FFAs also β-oxidize to generate acetyl coenzyme A (acetyl-CoA), which enters the tricarboxylic acid cycle and is oxidized to generate nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2). The electron transport system then uses the NADH and FADH2 to produce a proton gradient. BAT uniquely expresses the uncoupling protein, Ucp1, which dissipates the proton gradient across the inner mitochondrial membrane, resulting in inefficiency during the formation of ATP in oxidative phosphorylation (OXPHOS). When Ucp1 is induced in the BAT, such as during cold stress or thyroid hormone (TH) stimulation, mitochondrial respiration is stimulated to the maximum amount to compensate for the gradient loss, generating heat in the process [44,83-85,135,136].
Adipocytes express the insulin receptor and take up glucose via the insulin-stimulated glucose transporter type 4 (GLUT4) pathway, functioning as essential sinks of glucose, regulating systemic blood glucose levels. Selective deletion of the insulin receptor in UCP1-positive adipocytes has been reported to result in an age-dependent loss of interscapular BAT and systemic glucose intolerance in mice. Unlike white fat cells that solely rely on GLUT4 for glucose uptake, brown adipocytes can also take up glucose via the GLUT1 transporter in an adrenergic-dependent and insulin-independent manner. This mechanism involves the activity of nutrient sensor mTORC2 downstream of β3-AR signalling. The relative roles and modes of regulation of GLUT1 versus GLUT4-dependent glucose uptake in thermogenic fat in vivo will require further investigation [116,137-140].
Notably, cold acclimation profoundly enhances systemic BCAA clearance preferentially in human individuals with active thermogenic fat. Consistent with observations in humans, cold exposure significantly reduced plasma Val, Leu and Ile in mice, but this effect was absent in mice with ablated thermogenic adipocytes. This BCAA sink function is an essential aspect of thermogenic fat function in glucose homeostasis because defective BCAA oxidation leads to diet-induced weight gain, glucose intolerance and insulin resistance in mice. In line with this, increased circulating BCAA levels, owing to reduced BCAA clearance, are linked to obesity, insulin resistance and type 2 diabetes in humans [124,133,138,139].
Since BAT has a high mitochondrial content, it is metabolically active and prone to oxidative damage. Increases in the level of reduced glutathione and activities of superoxide dismutase, catalase, glutathione peroxidase, and glutathione reductase suggest there is an elevated level of reactive oxidative species (ROS) in rat BAT during cold acclimation.
UCP1 is located in the mitochondrial inner membrane and uncouples the proton (H+) gradient from ATP synthesis. UCP1 activity is inhibited by purine nucleotides, whereas long-chain free fatty acids, which are negatively charged at the carboxyl end, bind to UCP1 and trigger the transfer of H+ into the matrix as a fatty acid anion/H+ symporter. One of the mechanisms of UCP1-independent thermogenesis in beige adipocytes is futile Ca2+ cycling in and out of the endoplasmic reticulum. The Ca2+ cycle involves Ca2+ uptake into the endoplasmic reticulum by sarcoplasmic/endoplasmic reticulum calcium ATPase 2B (SERCA2B) and its release by ryanodine receptor 2 (RYR2) and inositol trisphosphate receptor (IP3R), which is coupled to ATP hydrolysis by SERCA2B and heat generation. Activation of the α1-adrenergic receptor (α1-AR) and β3-AR, in response to norepinephrine, triggers cellular Ca2+ flux and its futile cycling. Under certain conditions, such as increased cytosolic Ca2+ levels (for example, enhanced Ca2+ release from RYR2 or IP3R), ATP abundance (high ATP/ADP ratio) or reduced binding affinity of SERCA2 to Ca2+ (often regulated by micro peptides), ATP hydrolysis by SERCA2 is uncoupled from Ca2+ uptake, thereby being highly exothermic. Another UCP1-independent thermogenic mechanism is creatine substrate cycling, which involves ATP-dependent phosphorylation of creatine by mitochondria-localized creatine kinase (Mi-CK) to phosphocreatine (PCr) and PCr dephosphorylation by unknown diphosphatases (Enz1-n). Lipolysis of triglycerides (TAGs) generates glycerol and fatty acids, which can be re-esterified back to TAG (TAG-fatty acid cycling). This process involves ATP-dependent conversion of glycerol to glycerol 3-phosphate (G3P) by glycerol kinase (GyK). G3P is also a key component of the NADH-G3P shuttle, which involves interconversion of G3P and dihydroxyacetone phosphate (DHAP) and allows for rapid ATP synthesis in the mitochondria. This cycle is promoted by thiazolidinediones, which activate GyK and cold exposure and the satiety hormone leptin, which promote lipolysis. AAC, ADP/ATP carrier [125,139,141-144].
5-HT does not cross the blood-brain barrier, and peripheral and central 5-HT represent two distinct pools. 5-HT regulates, via the hypothalamus, brain stem and the spinal cord, satiety, hepatic glucose uptake and adaptation to cold exposure is regulated by central 5-HT. Peripheral 5-HT produced by intestinal enterochromaffin cells, pancreatic islets, and adipose tissue exerts local or systemic lipid and glucose homeostasis control through distinct 5-HT receptors.
Central 5-HT promotes thermogenesis since pharmacological or genetic depletion of central 5-HT has impaired thermogenic adaptation to cold. It is related central depletion of 5-HT have reduced adaptation to cold exposure, diminished thermogenic function of BAT, and decreased recruitment of beige adipocytes. Central 5-HT appears to increase BAT and beige adipocyte thermogenic function by modulating sympathetic outflow to these tissues. Transynaptic retrograde tracing from BAT synaptic terminals shows glutamatergic and 5-HT neurons of the rostral raphe pallidus synapse onto sympathetic fibres in the intermediolateral nucleus (IML) of the spinal cord. Systemic or IML injections of 5-HT or fenfluramine increase the sympathetic firing of these fibres. In contrast, IML injections of 5-HT7 receptor antagonists decrease sympathetic tone. Central 5-HT pathways play a significant role in adapting to cold exposure through the sympathetic activation of thermogenic adipose tissue [4,5,13,29,40,49,54,145-147].
The degradation of 5-HT is mainly catalyzed by mitochondrial monoamine oxidase A (MAO-A), generating 5-hydroxyindolic acid and reactive oxygen species (ROS), primarily H2O2. In addition to its role as a neurotransmitter in the regulation of central nervous system function, 5-HT also has multiple physiological functions in the periphery. 5-HT system, including 5-HT synthesis and 5-HT2 R, is activated in the hepatocytes of a T2DM mouse model, which crucially affected the occurrence of hepatic steatosis and inflammation with fibrosis. 5-HT2 R activation in hepatocytes regulates PKCε activation with the subsequent phosphorylation of Akt, mTOR, and ERK1/2, ultimately resulting in ELS, including de novo lipogenesis, and TG and VLDL synthesis with LDA, whereas the activation of both 5-HT synthesis and 5-HT2 R modulates oxidative stress with the activation of NF-κB and inflammatory signalling molecules, including p38, JNK, and STAT3 [31,40,124,144].
Serotonin promotes a fast stimulation in glucose uptake in both L6 myotubes and skeletal muscle mediated through the Htr2a receptor. The other thesis insists that incubation with serotonin-induced increased 2-deoxyglucose uptake in a concentration-dependent fashion by translocating GLUT4 to the cell membrane. GLUT4 translocation is caused by serotonylation of the small GTPase Rab4. In Serotonin signals 6-phosphofructo-1-kinase (PFK) through the Htr2a, the major rate-limiting enzyme of glycolysis and is related to the entire glycolytic pathway in the skeletal muscle.
Serotonin increases glucose uptake and glycolysis through Htr2a and intracellular serotonylation by stimulation of phospholipase C (PLC) in the skeletal muscle promotes the recruitment of protein kinase C (PKC) and calmodulin and the activation of calmodulin kinase II, which connects with PFK upon serotonin action [129,138,144,148,149].
Accumulation of the visceral WAT is highly associated with insulin resistance and diabetes. White adipose tissue (WAT) and brown adipose tissue (BAT) stores energy and generate body heat. WAT is stored subcutaneously and viscerally, surrounding intra-abdominal organs such as the liver, pancreas, and intestines.
Brown adipocytes have a common embryological origin with myocytes and are found mainly in humans' intrascapular, paraspinal, supraclavicular, suprarenal, pericardial, and para-aortic regions. Similar to muscle, it has a high concentration of mitochondria, which gives it a characteristic brown colour. The second population, known as "beige" or "bite" ("brown in white") adipocytes, is derived from a subpopulation of white adipocytes interspersed within the WAT. In humans, BAT is present at birth and regresses with age, although it remains metabolically active throughout adulthood. BAT is highly innervated and vascularized, rapidly responding to external stimulation such as cold and diet. BAT utilizes available substrates such as intracellular triacylglycerol (TAG) and blood glucose as fuels. Cold improves insulin sensitivity in patients with type 2 diabetes. Since cold acclimation can recruit the BAT in obese patients, BAT activation may also improve obesity-associated insulin resistance and hyperglycemia. The activity of adult brown and beige fat decreases with ageing and may contribute to the progression of chronic metabolic diseases. It appears that activating and recruiting brown and beige fat would be beneficial to improving overall metabolic health [124,125,133].
Thermogenesis in obese patients or patients with insulin resistance is something profound in patients infected with Sars-Cov-2. The mechanism proceeds as explained above. There is a 5-HT deficit due to Tryptophan malabsorption due to the internalization of ACE-2 in the intestine. There is the consumption of serum 5-HT, freeing space for the action of catecholamines, inducing UCP-1 in brown and beige adipocytes. Thermogenesis is one more stimulus to inflammation and the perpetuation of oxidative stress. Keeping the critically ill patient on high doses of norepinephrine amplifies this inflammatory pathway further, which is why it is necessary to associate vasopressin, trying to reduce the amount of norepinephrine supplied to sustain the blood pressure of the critically ill patient. Also, it is essential to remember that stress constantly stimulates cortisol and the release of adrenaline. However, these pathways are depleted due to the malabsorption of Tryptophan (serotonin) and Phenylalanine (tyrosine and catecholamines). Managing the patient COVID-19 is a great challenge due to the interference that the disease causes in multiple essential metabolic pathways in the control of immunity, inflammation and homeostasis (Figure 6, Figure 7, Figure 8, Figure 9, Figure 10, Figure 11, Figure 12, Figure 13, Figure 14, Figure 15, Figure 16, Figure 17 and Figure 18).
 Figure 6: Brown adipose tissue physiology. The activation of the sympathetic nervous system via cold results in the release of norepinephrine. The catecholamine binds to ß-adrenergic receptors (ß-AR) leading to the transcription of the central thermogenic factor uncoupling protein-1 (UCP1). UCP1, located in the mitochondria, uncouples respiration from the ATP synthesis and thus induces the generation of heat (thermogenesis). In addition to cold-stimulated sympathetic activation, endogenous factors (e.g., PGC1α, PRDM16, bone morphogenetic proteins, irisin, meteorin-like and FGF21) play an important role in the activation of a thermogenic phenotype; however, ß-adrenergic stimulation in white adipose tissue results primarily in lipolysis. TG: Triglycerides [169].
View Figure 6
Figure 6: Brown adipose tissue physiology. The activation of the sympathetic nervous system via cold results in the release of norepinephrine. The catecholamine binds to ß-adrenergic receptors (ß-AR) leading to the transcription of the central thermogenic factor uncoupling protein-1 (UCP1). UCP1, located in the mitochondria, uncouples respiration from the ATP synthesis and thus induces the generation of heat (thermogenesis). In addition to cold-stimulated sympathetic activation, endogenous factors (e.g., PGC1α, PRDM16, bone morphogenetic proteins, irisin, meteorin-like and FGF21) play an important role in the activation of a thermogenic phenotype; however, ß-adrenergic stimulation in white adipose tissue results primarily in lipolysis. TG: Triglycerides [169].
View Figure 6
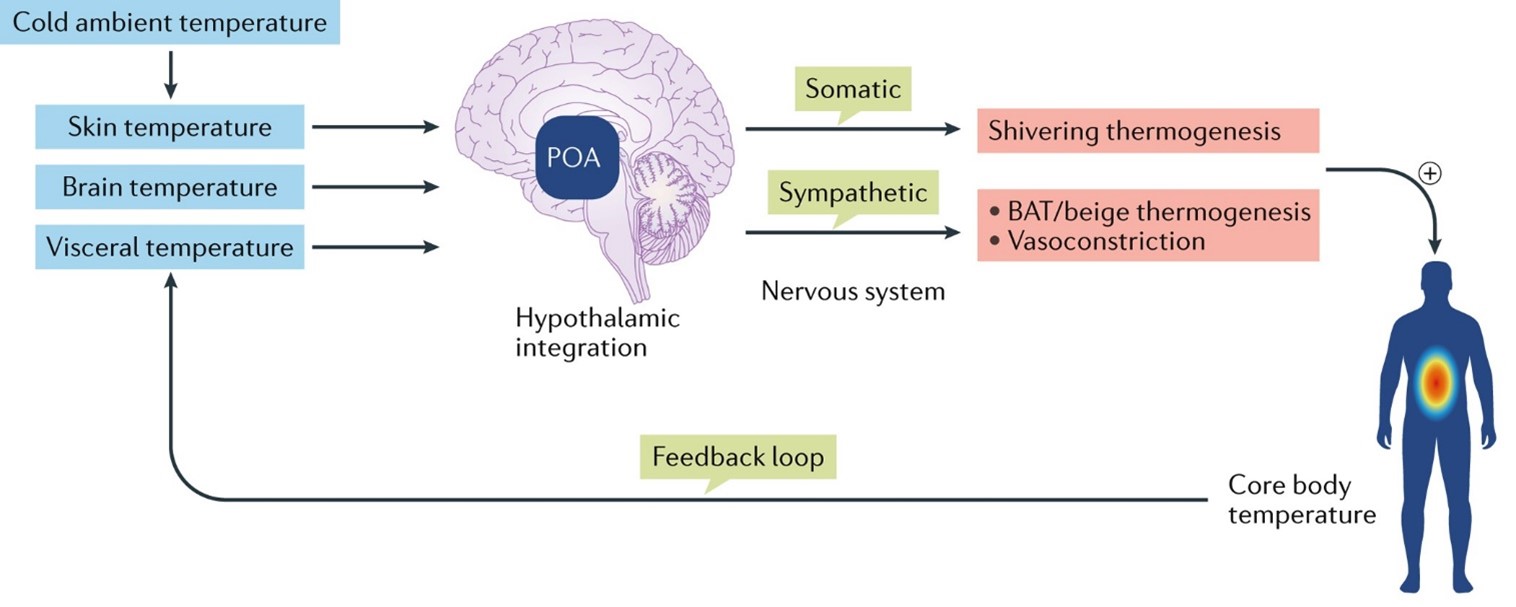 Figure 7: Modest cooling of the skin below a temperature of 26 °C activates transient receptor potential cation channel subfamily M member 8 (TRPM8) in sensory neurons (not shown), which acts as a sensor for mild, non-noxious cold and mediates the sensation of cold to the central nervous system. The signals from the skin are integrated in the preoptic area (POA) of the hypothalamus together with temperature sensations from the brain and viscera. Sympathetic and somatic nervous system outflow from the POA stimulates shivering as well as non-shivering thermogenesis, thus pre-emptively counteracting cold ambient temperatures. BAT is characterized by a dense vasculature and sympathoadrenergic innervation that differs markedly from that of WAT. White adipocytes contain a single large lipid droplet surrounded by a narrow rim of cytosol and few mitochondria. By contrast, brown adipocytes store lipids in multiple small droplets and contain large numbers of mitochondria. A unique feature of BAT mitochondria is the presence of UCP1 in the inner mitochondrial membrane. UCP1 is a protonophore that can uncouple oxidative phosphorylation from ATP regeneration, thereby converting energy stored in the form of triglycerides into heat BAT. The brown adipocytes found in the interscapular depot are known as 'classic brown adipocytes'. Although classic BAT constitutively expresses a high level of UCP1 even at high ambient temperatures, UCP1 levels are low in beige fat depots unless they are stimulated by cold or catecholamines, which increase UCP1 levels dramatically to those found in classic BA. Whereas the transcriptional signature of brown and beige adipocytes differs and reflects the different origin of the cells 45,46, the morphology and especially the thermogenic capacity are similar in classic brown and beige adipocytes 47,48; several crucial transcription factors such as peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α) and PR domain zinc-finger protein 16 (PRDM16) are common to both classic brown and beige adipocytes brown adipose tissue [128].
View Figure 7
Figure 7: Modest cooling of the skin below a temperature of 26 °C activates transient receptor potential cation channel subfamily M member 8 (TRPM8) in sensory neurons (not shown), which acts as a sensor for mild, non-noxious cold and mediates the sensation of cold to the central nervous system. The signals from the skin are integrated in the preoptic area (POA) of the hypothalamus together with temperature sensations from the brain and viscera. Sympathetic and somatic nervous system outflow from the POA stimulates shivering as well as non-shivering thermogenesis, thus pre-emptively counteracting cold ambient temperatures. BAT is characterized by a dense vasculature and sympathoadrenergic innervation that differs markedly from that of WAT. White adipocytes contain a single large lipid droplet surrounded by a narrow rim of cytosol and few mitochondria. By contrast, brown adipocytes store lipids in multiple small droplets and contain large numbers of mitochondria. A unique feature of BAT mitochondria is the presence of UCP1 in the inner mitochondrial membrane. UCP1 is a protonophore that can uncouple oxidative phosphorylation from ATP regeneration, thereby converting energy stored in the form of triglycerides into heat BAT. The brown adipocytes found in the interscapular depot are known as 'classic brown adipocytes'. Although classic BAT constitutively expresses a high level of UCP1 even at high ambient temperatures, UCP1 levels are low in beige fat depots unless they are stimulated by cold or catecholamines, which increase UCP1 levels dramatically to those found in classic BA. Whereas the transcriptional signature of brown and beige adipocytes differs and reflects the different origin of the cells 45,46, the morphology and especially the thermogenic capacity are similar in classic brown and beige adipocytes 47,48; several crucial transcription factors such as peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α) and PR domain zinc-finger protein 16 (PRDM16) are common to both classic brown and beige adipocytes brown adipose tissue [128].
View Figure 7
 Figure 8: Uncoupling protein 1 (UCP1). The respiratory chain pumps protons into the intermembrane space, which creates a gradient whose electromotive force drives ATP synthase. In brown adipocytes, this gradient can be short-circuited by activation of UCP1, which dissipates the energy stored in the gradient as heat. b | Creatine cycling (conceptual model). Mitochondrial creatine kinase (CKMT) transfers an activated phosphate from ATP to creatine (Cr), which forms phosphor-creatine (P-Cr). The resulting ADP is transferred into the mitochondrial matrix and drives oxidative phosphorylation. The P-Cr is hydrolysed by P-Cr-ase and is available for another futile cycle. c | Sarcolipin (SLN) and sarcoplasmic/endoplasmic reticulum calcium ATPases (SERCAs). SLN (a small transmembrane proteolipid) reduces the efficiency of SERCAs, which leads to increased availability of ADP and thus increased activity of oxidative phosphorylation and the tricarboxylic acid (TCA) cycle. This is probably an important mechanism of non-shivering thermogenesis in muscle. Among the emerging mechanisms implicated in thermogenesis, creatine cycling has recently been reported to contribute markedly to thermogenesis in beige adipocytes91. Earlier work had already detected an increase in mitochondrial-type creatine kinase in BAT of cold-exposed rats92. Using proteomic analysis of mitochondria, Kazak et al. identified arginine-creatine metabolism as a specific signature of beige adipocytes in mice91. Short-term cold exposure (6 h) dramatically increased gene expression and protein levels of creatine kinase U-type, mitochondrial (CKMT1) and creatine kinase S-type, mitochondrial (CKMT2) primarily in beige adipose tissue. Moreover, the addition of creatine to mitochondria from beige adipocytes stimulated respiration when levels of ADP were limiting respiration. This effect was not seen in mitochondria from muscle or brown adipocytes. In vivo inhibition of creatine transport reduced catecholamine-stimulated whole-body oxygen consumption by decreasing beige adipose tissue respiration91. Moreover, Ckmt1 gene expression was upregulated in the beige adipose tissue of Ucp1-knockout mice, which indicates a compensatory role for creatine-cycling-mediated thermogenesis in the absence of UCP1. Blocking creatine transport in cultured human brown adipocytes nearly halved oxygen consumption, and small interfering RNA-mediated knockdown of Ckmt1 also diminished respiration Cyt c, cytochrome c; RYR1, ryanodine receptor 1 [128].
View Figure 8
Figure 8: Uncoupling protein 1 (UCP1). The respiratory chain pumps protons into the intermembrane space, which creates a gradient whose electromotive force drives ATP synthase. In brown adipocytes, this gradient can be short-circuited by activation of UCP1, which dissipates the energy stored in the gradient as heat. b | Creatine cycling (conceptual model). Mitochondrial creatine kinase (CKMT) transfers an activated phosphate from ATP to creatine (Cr), which forms phosphor-creatine (P-Cr). The resulting ADP is transferred into the mitochondrial matrix and drives oxidative phosphorylation. The P-Cr is hydrolysed by P-Cr-ase and is available for another futile cycle. c | Sarcolipin (SLN) and sarcoplasmic/endoplasmic reticulum calcium ATPases (SERCAs). SLN (a small transmembrane proteolipid) reduces the efficiency of SERCAs, which leads to increased availability of ADP and thus increased activity of oxidative phosphorylation and the tricarboxylic acid (TCA) cycle. This is probably an important mechanism of non-shivering thermogenesis in muscle. Among the emerging mechanisms implicated in thermogenesis, creatine cycling has recently been reported to contribute markedly to thermogenesis in beige adipocytes91. Earlier work had already detected an increase in mitochondrial-type creatine kinase in BAT of cold-exposed rats92. Using proteomic analysis of mitochondria, Kazak et al. identified arginine-creatine metabolism as a specific signature of beige adipocytes in mice91. Short-term cold exposure (6 h) dramatically increased gene expression and protein levels of creatine kinase U-type, mitochondrial (CKMT1) and creatine kinase S-type, mitochondrial (CKMT2) primarily in beige adipose tissue. Moreover, the addition of creatine to mitochondria from beige adipocytes stimulated respiration when levels of ADP were limiting respiration. This effect was not seen in mitochondria from muscle or brown adipocytes. In vivo inhibition of creatine transport reduced catecholamine-stimulated whole-body oxygen consumption by decreasing beige adipose tissue respiration91. Moreover, Ckmt1 gene expression was upregulated in the beige adipose tissue of Ucp1-knockout mice, which indicates a compensatory role for creatine-cycling-mediated thermogenesis in the absence of UCP1. Blocking creatine transport in cultured human brown adipocytes nearly halved oxygen consumption, and small interfering RNA-mediated knockdown of Ckmt1 also diminished respiration Cyt c, cytochrome c; RYR1, ryanodine receptor 1 [128].
View Figure 8
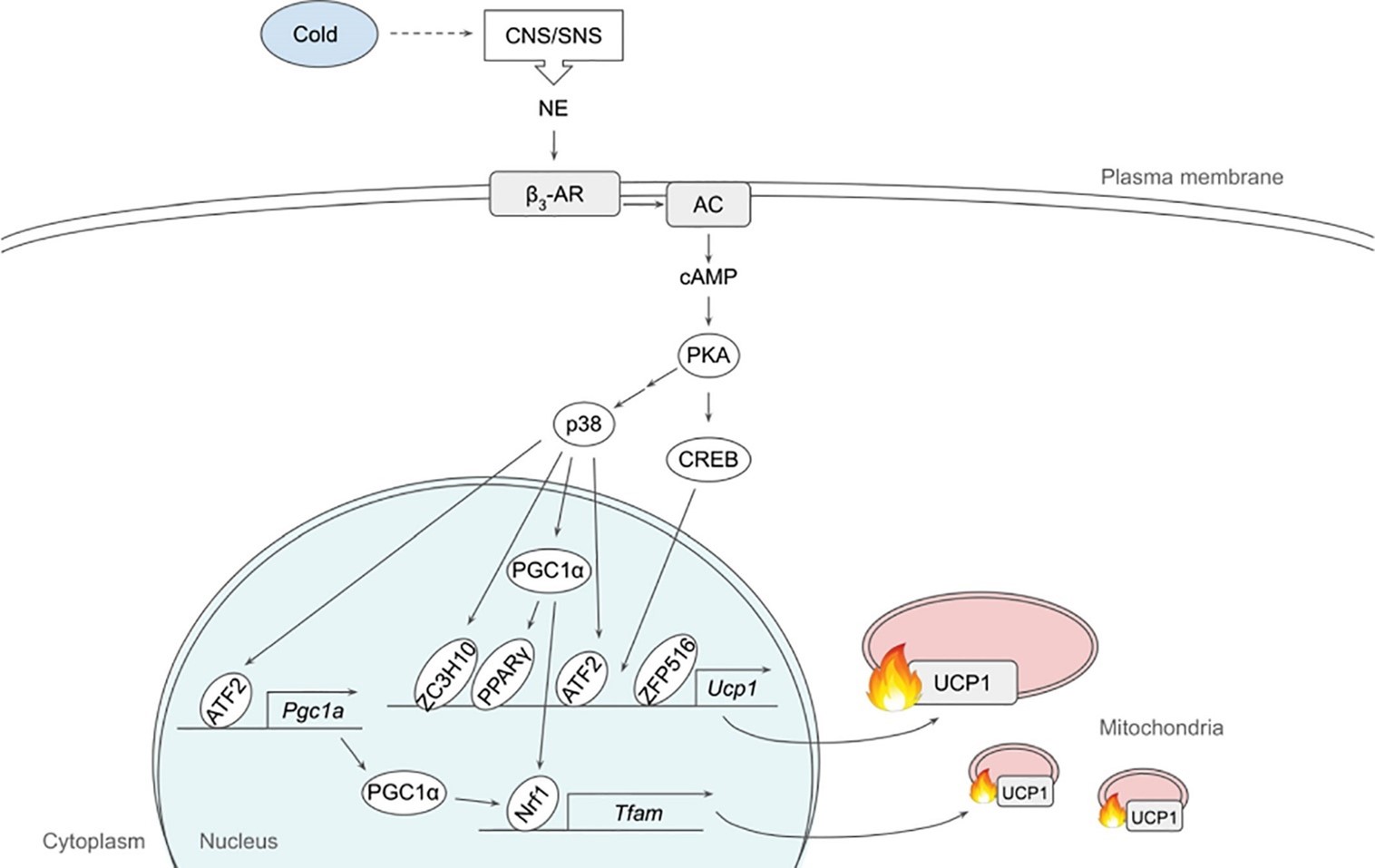 Figure 9: Schematic model of β3-adrenergic signalling pathway that promotes thermogenesis in adipocytes. Cold stimulates CNS/SNS to secrete NE that binds β3-AR, which then activates AC producing cAMP. cAMP in turn activates PKA that has a variety of downstream targets, including transcription factors to upregulate thermogenic gene expression. See text for details. AC, adenylate cyclase; ATF2, activating transcription factor 2; β3-AR, β3-adrenergic receptor; CNS, central nervous system; CREB: cAMP response-element binding protein; ETC: Electron Transport Chain; FFA: Free Fatty Acid; IRF4: Interferon Regulatory Factor 4; NE: Norepinephrine; NRF1: Nuclear Respiratory Factor 1; PGC1α: Peroxisome Proliferator Activated Receptor γ coactivator α; PKA: Protein Kinase A; PPARγ: Peroxisome Proliferator Activated Receptor γ; SNS: Sympathetic Nervous System; TCA: Tricarboxylic Acid; TFAM: Mitochondrial Transcription Factor A; UCP1: Uncoupling Protein 1; ZC3H10: Zinc Finger CCCH-type containing 10; ZFP516: Zinc Finger Protein 516 [125].
View Figure 9
Figure 9: Schematic model of β3-adrenergic signalling pathway that promotes thermogenesis in adipocytes. Cold stimulates CNS/SNS to secrete NE that binds β3-AR, which then activates AC producing cAMP. cAMP in turn activates PKA that has a variety of downstream targets, including transcription factors to upregulate thermogenic gene expression. See text for details. AC, adenylate cyclase; ATF2, activating transcription factor 2; β3-AR, β3-adrenergic receptor; CNS, central nervous system; CREB: cAMP response-element binding protein; ETC: Electron Transport Chain; FFA: Free Fatty Acid; IRF4: Interferon Regulatory Factor 4; NE: Norepinephrine; NRF1: Nuclear Respiratory Factor 1; PGC1α: Peroxisome Proliferator Activated Receptor γ coactivator α; PKA: Protein Kinase A; PPARγ: Peroxisome Proliferator Activated Receptor γ; SNS: Sympathetic Nervous System; TCA: Tricarboxylic Acid; TFAM: Mitochondrial Transcription Factor A; UCP1: Uncoupling Protein 1; ZC3H10: Zinc Finger CCCH-type containing 10; ZFP516: Zinc Finger Protein 516 [125].
View Figure 9
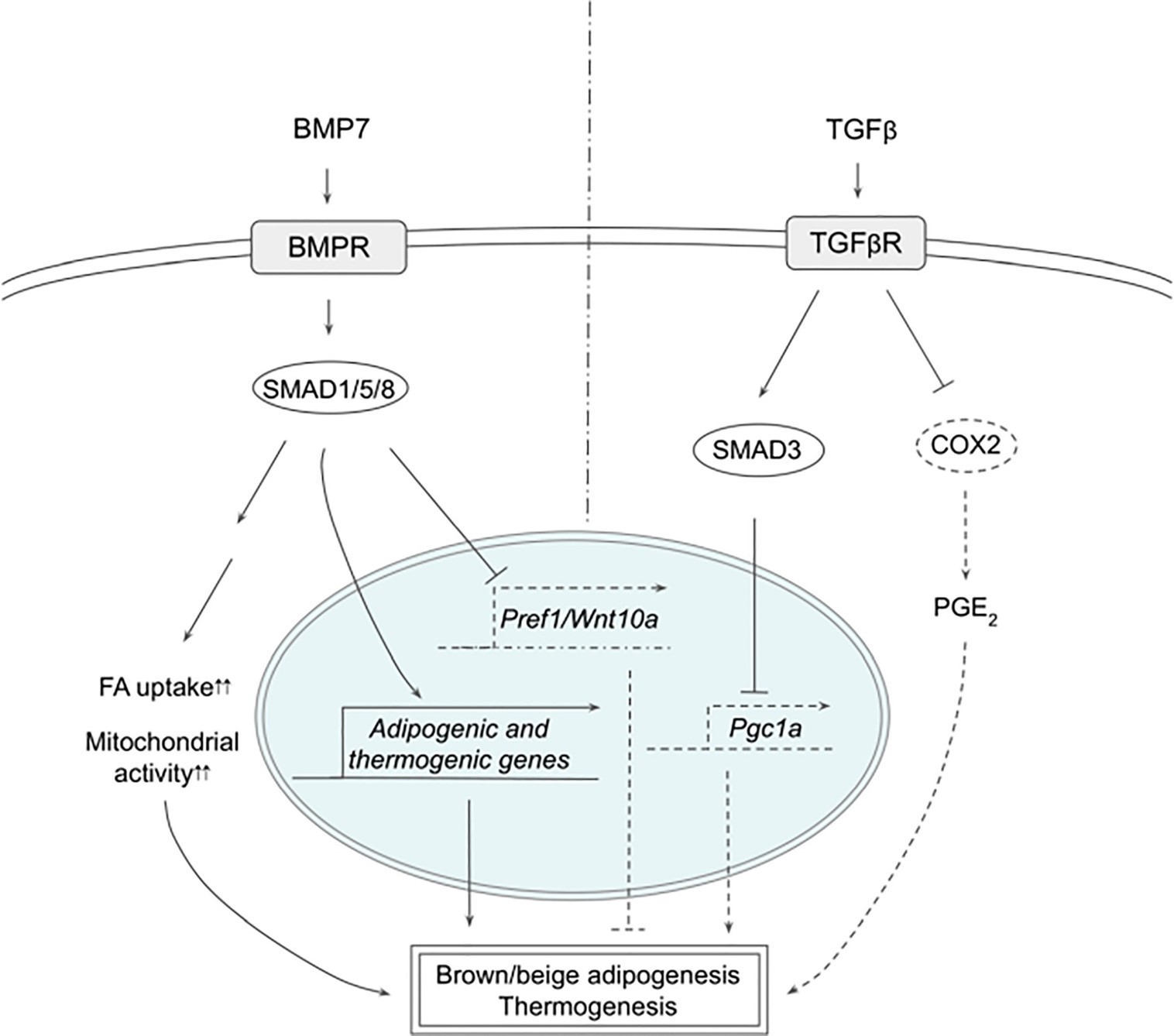 Figure 10: Schematic models of BMP7 and TGFβ signalling pathways in thermogenesis. BMP7 binds TGFβR, which activates SMAD1/5/8, leading to expression of adipogenic and thermogenic genes as well as suppression of Pref1 and Wnt10a in precursor cells to promote brown and beige adipogenesis. In mature adipocytes, BMP7 signalling increases FA uptake and mitochondrial activity, resulting in enhanced thermogenesis. TGFβ activates SMAD2/3 that suppresses PGC1α expression and COX2/PGE2 pathway to reduce thermogenesis. BMP7 induced the expression of both adipogenic and brown-adipocyte specific genes, such as CEBPs, PPARγ, PRDM16, PGC1α, and UCP1, while downregulating adipogenic suppressors such as Pref-1 and WNT10A. BMP7 KO mice exhibited reduced BAT mass that had almost undetectable UCP1 expression. In contrast, BMP7 overexpression via adenovirus injection increased the expression of PRDM16 and UCP1 in BAT, energy expenditure and body temperature, while decreasing body weight. In mature brown adipocytes, BMP7 increased mitochondrial activity through p38-ATF2 and SMAD1/5/8 pathways. BMP7-treated cells upregulated the expression of CD36 and CPT1 that transport FAs into the cells and mitochondria, respectively, resulting in increased citrate synthase activity and FA oxidation. Administration of BMP7 in vivo increased the expression of CD36 in BAT, which was accompanied by increased oxygen consumption. In addition, respiratory exchange ratio in these animals decreased, reflecting the substrate utilization switching towards fatty acids. Another study also confirmed the similar effects of BMP7, where BMP7 administration increased the expression of UCP1 and CD36 in BAT, BAT mass, energy expenditure and fat oxidation. Furthermore, these authors found that BMP7 induced the expression of BAT genes in WAT depots with overall reduced WAT size, suggesting that BMP7 promotes browning of WAT. This is supported by a finding that demonstrated BMP7 driving human adipogenic stem cells into beige adipocytes. BMP7 treatment promoted the expression of beige markers, such as CD137 and TMEM26, in addition to the expression of thermogenic genes, such as UCP1, CIDEA and DIO2 BMP7, bone morphogenetic protein 7; COX2: Cyclooxygenase 2; FA: Fatty Acid; PGC1α: Peroxisome Proliferator Activated Receptor γ; PGE2: Prostaglandin E2; PREF1: Preadipocyte Factor 1; SMAD: Mothers Against Decapentaplegic Homolog; TβR1: TGFβ Receptor 1; TGFβ: Transforming Growth Factor Beta; TGFβR: TGFβ Receptor; WNT10a: Wnt Family Member 10A [125].
View Figure 10
Figure 10: Schematic models of BMP7 and TGFβ signalling pathways in thermogenesis. BMP7 binds TGFβR, which activates SMAD1/5/8, leading to expression of adipogenic and thermogenic genes as well as suppression of Pref1 and Wnt10a in precursor cells to promote brown and beige adipogenesis. In mature adipocytes, BMP7 signalling increases FA uptake and mitochondrial activity, resulting in enhanced thermogenesis. TGFβ activates SMAD2/3 that suppresses PGC1α expression and COX2/PGE2 pathway to reduce thermogenesis. BMP7 induced the expression of both adipogenic and brown-adipocyte specific genes, such as CEBPs, PPARγ, PRDM16, PGC1α, and UCP1, while downregulating adipogenic suppressors such as Pref-1 and WNT10A. BMP7 KO mice exhibited reduced BAT mass that had almost undetectable UCP1 expression. In contrast, BMP7 overexpression via adenovirus injection increased the expression of PRDM16 and UCP1 in BAT, energy expenditure and body temperature, while decreasing body weight. In mature brown adipocytes, BMP7 increased mitochondrial activity through p38-ATF2 and SMAD1/5/8 pathways. BMP7-treated cells upregulated the expression of CD36 and CPT1 that transport FAs into the cells and mitochondria, respectively, resulting in increased citrate synthase activity and FA oxidation. Administration of BMP7 in vivo increased the expression of CD36 in BAT, which was accompanied by increased oxygen consumption. In addition, respiratory exchange ratio in these animals decreased, reflecting the substrate utilization switching towards fatty acids. Another study also confirmed the similar effects of BMP7, where BMP7 administration increased the expression of UCP1 and CD36 in BAT, BAT mass, energy expenditure and fat oxidation. Furthermore, these authors found that BMP7 induced the expression of BAT genes in WAT depots with overall reduced WAT size, suggesting that BMP7 promotes browning of WAT. This is supported by a finding that demonstrated BMP7 driving human adipogenic stem cells into beige adipocytes. BMP7 treatment promoted the expression of beige markers, such as CD137 and TMEM26, in addition to the expression of thermogenic genes, such as UCP1, CIDEA and DIO2 BMP7, bone morphogenetic protein 7; COX2: Cyclooxygenase 2; FA: Fatty Acid; PGC1α: Peroxisome Proliferator Activated Receptor γ; PGE2: Prostaglandin E2; PREF1: Preadipocyte Factor 1; SMAD: Mothers Against Decapentaplegic Homolog; TβR1: TGFβ Receptor 1; TGFβ: Transforming Growth Factor Beta; TGFβR: TGFβ Receptor; WNT10a: Wnt Family Member 10A [125].
View Figure 10
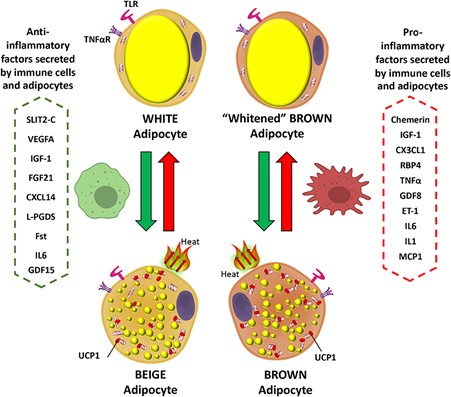 Figure 11: Inflammatory mediator actions on white and brown adipocytes. Pro-inflammatory factors secreted by immune cells and brown/beige adipocytes prevent the expression of brown fat genes in adipocytes, including UCP1, the main thermogenic protein (red arrows). In contrast anti-inflammatory mediators promote the transition of white to beige adipocytes and could prevent expression of the "whitened" brown adipocyte phenotype in brown adipose tissue (green arrows). Pro-inflammatory signalling can disrupt the insulin signalling cascade and impair the insulin sensitivity of BAT. Although, IL-1, TNF-α, MIF, and IL-6 have consistently been shown to cause insulin resistance in WAT, their effects have not been extensively explored in BAT at the molecular level. Elevated inflammatory marker levels in the diet induced obesity state in mice are suggested to be responsible for BAT insulin resistance via AKT (protein kinase B) and ERK pathways. TNF-α appears to play an important role in impairing insulin sensitivity of BAT. The mechanism has been discussed in detail and it involves disturbances of both MAP- kinases activation and IRS-2 and AKT. Mammalian target of rapamycin complex 2 (mTORC2), which activates inflammation, sustains thermogenesis via Akt-induced glucose uptake and glycolysis in BAT. TNF-α most dramatically alters 3T3-L1 adipocyte mitochondrial functions, whereas IL-1β and IL-6 have more modest effects. Moreover, activation of the NLRP3 inflammasome in macrophages attenuates UCP1 induction and mitochondrial respiration in cultures of primary adipocytes possibly via IL-1, while the absence of NLRP3 is protective for UCP1 and adaptive thermogenesis capacity in adipocytes IGF-1, Insulin-Like Growth Factor-1; CX3CL1, Fractaline; RBP4, Retinol-Binding Protein 4; TNFα, Tumor necrosis factor a; GDF8, Growth differentiation factor 8; ET-1, Endothelin 1; IL6, Interleukin 6; IL1, Interleukin 1; MCP1, Monocyte Chemoattractant Protein-1; SLIT2-C, C-terminal fragment of SLIT2 protein; VEGFA, Vascular endothelial growth factor A; FGF21, Fibroblast growth factor 21; CXCL14, C-X-C motif chemokine ligand-14; L-PGDS, Lipocalin prostaglandin D synthase; Fst, Follistatin; UCP1, Uncoupling Protein 1; GDF15, Growth and differentiation factor 15 [170].
View Figure 11
Figure 11: Inflammatory mediator actions on white and brown adipocytes. Pro-inflammatory factors secreted by immune cells and brown/beige adipocytes prevent the expression of brown fat genes in adipocytes, including UCP1, the main thermogenic protein (red arrows). In contrast anti-inflammatory mediators promote the transition of white to beige adipocytes and could prevent expression of the "whitened" brown adipocyte phenotype in brown adipose tissue (green arrows). Pro-inflammatory signalling can disrupt the insulin signalling cascade and impair the insulin sensitivity of BAT. Although, IL-1, TNF-α, MIF, and IL-6 have consistently been shown to cause insulin resistance in WAT, their effects have not been extensively explored in BAT at the molecular level. Elevated inflammatory marker levels in the diet induced obesity state in mice are suggested to be responsible for BAT insulin resistance via AKT (protein kinase B) and ERK pathways. TNF-α appears to play an important role in impairing insulin sensitivity of BAT. The mechanism has been discussed in detail and it involves disturbances of both MAP- kinases activation and IRS-2 and AKT. Mammalian target of rapamycin complex 2 (mTORC2), which activates inflammation, sustains thermogenesis via Akt-induced glucose uptake and glycolysis in BAT. TNF-α most dramatically alters 3T3-L1 adipocyte mitochondrial functions, whereas IL-1β and IL-6 have more modest effects. Moreover, activation of the NLRP3 inflammasome in macrophages attenuates UCP1 induction and mitochondrial respiration in cultures of primary adipocytes possibly via IL-1, while the absence of NLRP3 is protective for UCP1 and adaptive thermogenesis capacity in adipocytes IGF-1, Insulin-Like Growth Factor-1; CX3CL1, Fractaline; RBP4, Retinol-Binding Protein 4; TNFα, Tumor necrosis factor a; GDF8, Growth differentiation factor 8; ET-1, Endothelin 1; IL6, Interleukin 6; IL1, Interleukin 1; MCP1, Monocyte Chemoattractant Protein-1; SLIT2-C, C-terminal fragment of SLIT2 protein; VEGFA, Vascular endothelial growth factor A; FGF21, Fibroblast growth factor 21; CXCL14, C-X-C motif chemokine ligand-14; L-PGDS, Lipocalin prostaglandin D synthase; Fst, Follistatin; UCP1, Uncoupling Protein 1; GDF15, Growth and differentiation factor 15 [170].
View Figure 11
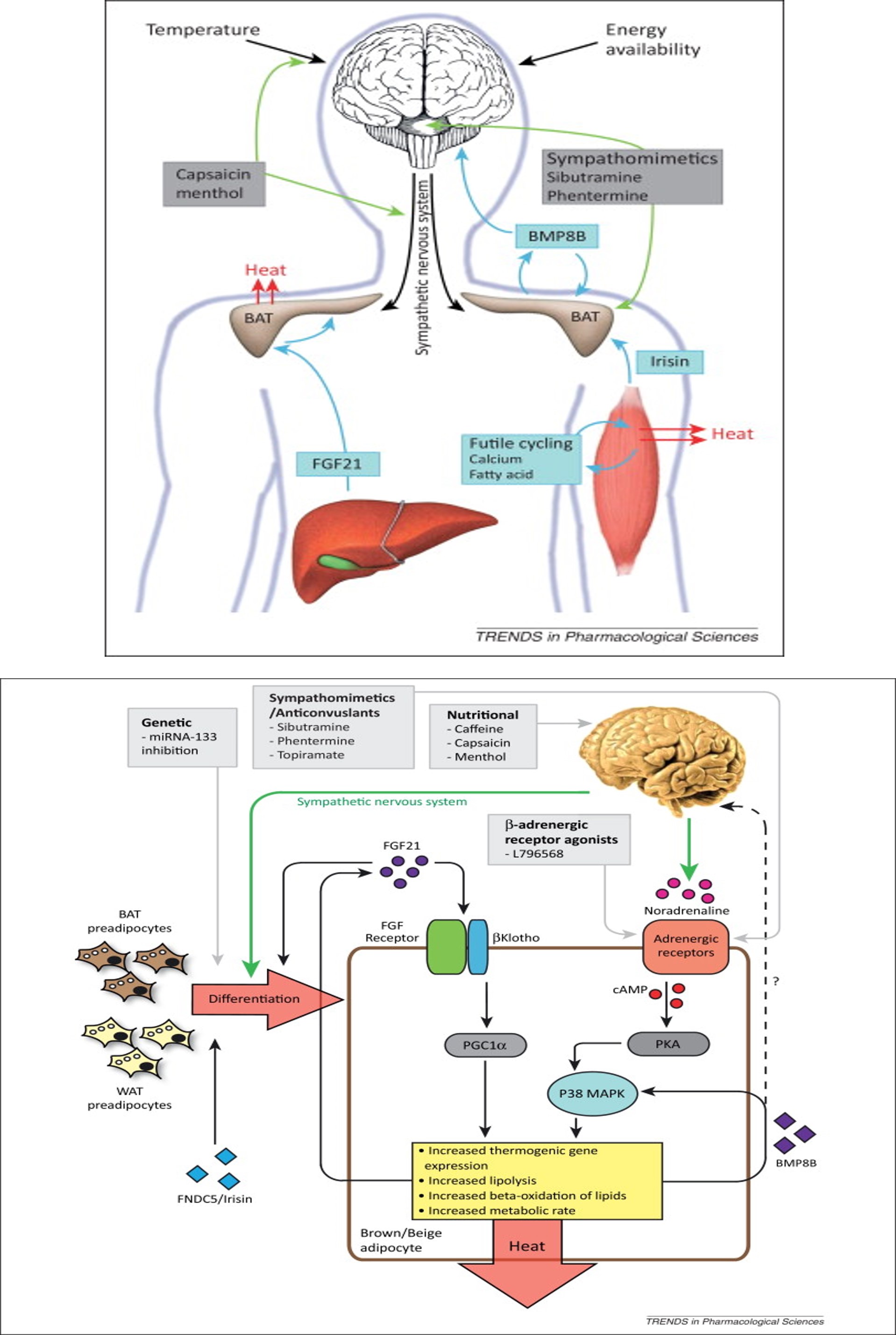 Figure 12: Biological and pharmacological activators of brown adipose tissue (BAT): inter-organ communication. Schematic showing the location of BAT and the broad range of tissues that signal to increase thermogenesis therein. The sympathetic nervous system is the primary signal to activate thermogenesis and recruitment of BAT. Blue boxes indicate endogenous molecules that modulate the effects of this activation either by co-ordinately activating thermogenic pathways or directly modifying central sympathetic outflow. Grey boxes indicate points at which pharmacological compounds can interact with and increase the activity of the biological pathways Molecular pathways regulating thermogenesis in brown adipose tissue (BAT). Diagram showing multiple signalling mechanisms that contribute positively to the overall thermogenic capacity of an organism. Black arrows define physiological pathways, grey arrows show the mechanism of action of various compounds known or suggested to increase thermogenesis. Green arrows indicate the importance of sympathetic nervous system activation for both recruitment of BAT and thermogenic activation [146].
View Figure 12
Figure 12: Biological and pharmacological activators of brown adipose tissue (BAT): inter-organ communication. Schematic showing the location of BAT and the broad range of tissues that signal to increase thermogenesis therein. The sympathetic nervous system is the primary signal to activate thermogenesis and recruitment of BAT. Blue boxes indicate endogenous molecules that modulate the effects of this activation either by co-ordinately activating thermogenic pathways or directly modifying central sympathetic outflow. Grey boxes indicate points at which pharmacological compounds can interact with and increase the activity of the biological pathways Molecular pathways regulating thermogenesis in brown adipose tissue (BAT). Diagram showing multiple signalling mechanisms that contribute positively to the overall thermogenic capacity of an organism. Black arrows define physiological pathways, grey arrows show the mechanism of action of various compounds known or suggested to increase thermogenesis. Green arrows indicate the importance of sympathetic nervous system activation for both recruitment of BAT and thermogenic activation [146].
View Figure 12
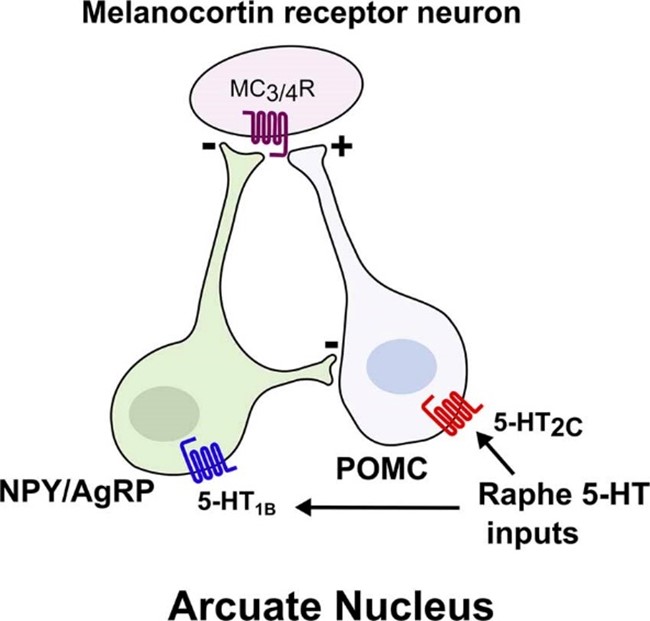 Figure 13: Brain 5-HT acts on central melanocortin neurons to suppress food intake. Melanocortin receptor neurons (MC3/4R) integrate signals from two reciprocal populations of neurons within the arcuate nucleus to promote satiety. Activation of 5-HT2c increases the activity of anorexigenic proopiomelanocortin (POMC) neurons, while activation of 5-HT1B inhibits orexigenic NPY/AgRP neurons. 5-HT's effects on energy balance, most notably the central melanocortin system, which includes two reciprocal populations of melanocortin neurons within the arcuate nucleus of the hypothalamus (ARC), anorexigenic neurons expressing proopiomelanocortin (POMC) and orexigenic neurons expressing neuropeptide Y/Agouti related peptide (NPY/AgRP). Melanocortin receptors (MC3R and MC4R) in downstream neurons such as the paraventricular nucleus (PVN) are activated by alpha-melanocyte stimulating hormone (α-MSH), a proteolytic product of POMC, and inhibited by AgRP to reciprocally regulate food intake and glucose homeostasis. Approximately 25% of POMC neurons in the adult mouse brain functionally express 5-HT2C receptors. 5-HT2C receptor activation in POMC neurons both induces Pomc mRNA expression and increases POMC neuronal activity through activation of TRPC5 cation channels. Remarkably, re-expression of 5-HT2C only in POMC neurons in an otherwise Htr2c−/− mouse is sufficient to reverse the hyperphagia and liver insulin resistance characteristic of Htr2c deficiency). Conversely, mice with a POMC neuron-specific deletion of Htr2c are hyperphagic, show a blunted anorectic response to fenfluramine or mCPP, and have impaired glucose homeostasis. Together, these studies underscore a critical role for 5-HT2C in POMC neurons to regulate food intake and hepatic glucose metabolism.
View Figure 13
Figure 13: Brain 5-HT acts on central melanocortin neurons to suppress food intake. Melanocortin receptor neurons (MC3/4R) integrate signals from two reciprocal populations of neurons within the arcuate nucleus to promote satiety. Activation of 5-HT2c increases the activity of anorexigenic proopiomelanocortin (POMC) neurons, while activation of 5-HT1B inhibits orexigenic NPY/AgRP neurons. 5-HT's effects on energy balance, most notably the central melanocortin system, which includes two reciprocal populations of melanocortin neurons within the arcuate nucleus of the hypothalamus (ARC), anorexigenic neurons expressing proopiomelanocortin (POMC) and orexigenic neurons expressing neuropeptide Y/Agouti related peptide (NPY/AgRP). Melanocortin receptors (MC3R and MC4R) in downstream neurons such as the paraventricular nucleus (PVN) are activated by alpha-melanocyte stimulating hormone (α-MSH), a proteolytic product of POMC, and inhibited by AgRP to reciprocally regulate food intake and glucose homeostasis. Approximately 25% of POMC neurons in the adult mouse brain functionally express 5-HT2C receptors. 5-HT2C receptor activation in POMC neurons both induces Pomc mRNA expression and increases POMC neuronal activity through activation of TRPC5 cation channels. Remarkably, re-expression of 5-HT2C only in POMC neurons in an otherwise Htr2c−/− mouse is sufficient to reverse the hyperphagia and liver insulin resistance characteristic of Htr2c deficiency). Conversely, mice with a POMC neuron-specific deletion of Htr2c are hyperphagic, show a blunted anorectic response to fenfluramine or mCPP, and have impaired glucose homeostasis. Together, these studies underscore a critical role for 5-HT2C in POMC neurons to regulate food intake and hepatic glucose metabolism.
View Figure 13
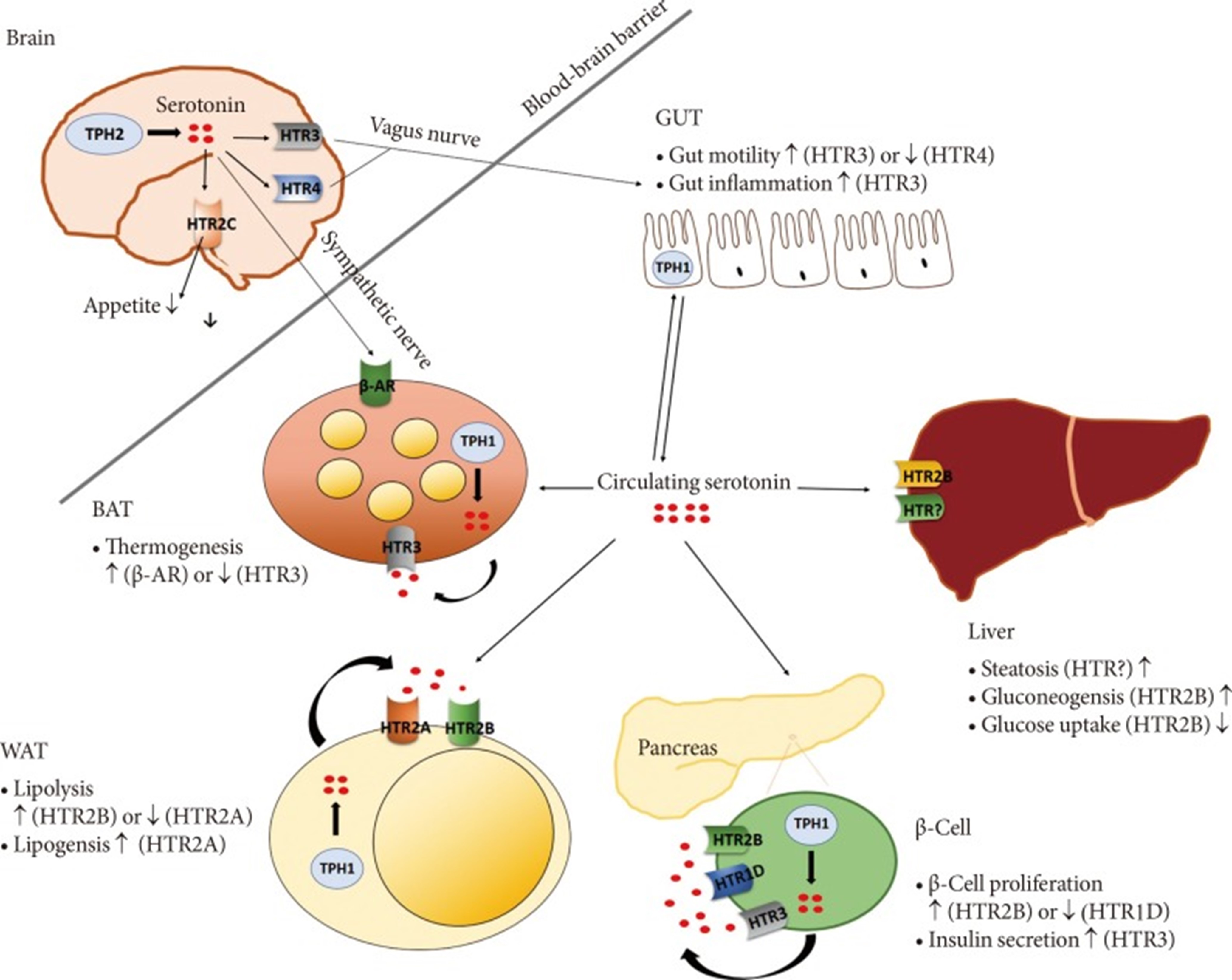 Figure 14: Functions of 5-hydroxytryptamine (5-HT) in metabolic organs. Central 5-HT regulates appetite and controls metabolic organs via the sympathetic/parasympathetic nervous system. Peripheral 5-HT regulates glucose metabolism, insulin resistance and energy expenditure in an autocrine/paracrine or endocrine manner. TPH, tryptophan hydroxylase; HTR, 5-HT receptor; β-AR β-adrenergic receptor; BAT, brown adipose tissue; WAT, white adipose tissue.
View Figure 14
Figure 14: Functions of 5-hydroxytryptamine (5-HT) in metabolic organs. Central 5-HT regulates appetite and controls metabolic organs via the sympathetic/parasympathetic nervous system. Peripheral 5-HT regulates glucose metabolism, insulin resistance and energy expenditure in an autocrine/paracrine or endocrine manner. TPH, tryptophan hydroxylase; HTR, 5-HT receptor; β-AR β-adrenergic receptor; BAT, brown adipose tissue; WAT, white adipose tissue.
View Figure 14
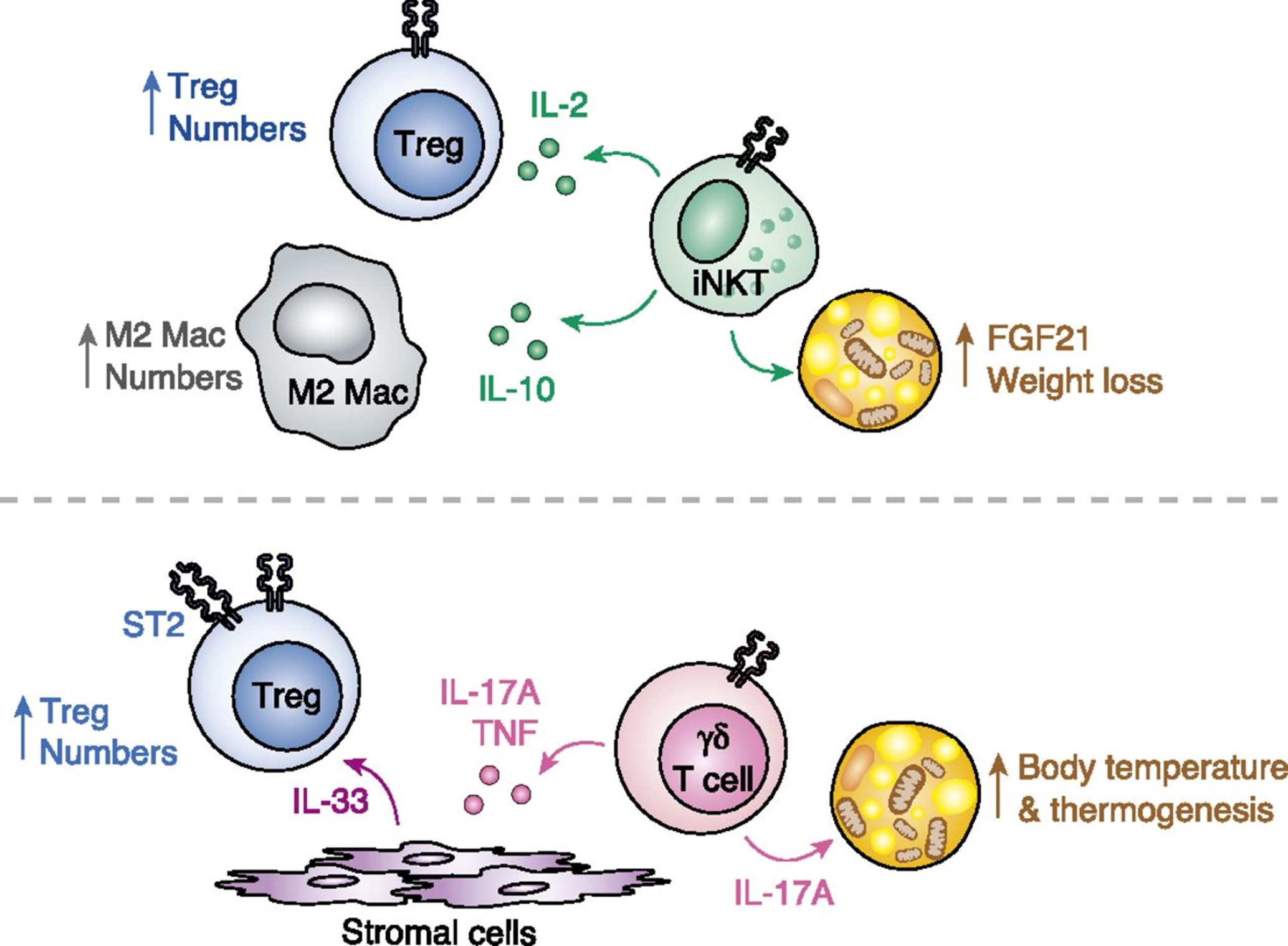 Figure 15: Upper figure. Cellular changes in adipose tissue in young and aged mice. Young adipose tissue is highly enriched with iNKT cells, which support M2 macrophage and Treg expansion by production of IL-10 and IL-2, respectively. As mice age, iNKT cells are gradually replaced by a population of IL-17A-secreting γδ T cells that support Tregs by a stromal cell-IL-33 axis. Lower figure. Nonredundant actions of iNKT and γδ T cells promote adipose tissue homeostasis. Top panel, Adipose iNKT cells expand Tregs via production of IL-2 and M2 macrophages via production of IL-10. Furthermore, activation of adipose iNKT cells induces weight loss and thermogenesis through an FGF21-dependent mechanism. Bottom panel, IL-17A-producing γδ T cells in adipose tissue drive Treg expansion by inducing stromal cells to produce IL-33. Additionally, a γδ IL-17A axis in adipose tissue is critical for maintenance of body temperature at thermoneutrality and upon cold challenge. iNKT cells and γδ cells, two innate T lymphocytes, have emerged as key regulators of immune homeostasis in visceral adipose tissue, as well as rheostats controlling body temperature in response to environmental fluctuations in sc and brown adipose depots (Figure 2). iNKT cells control adipose Tregs and macrophage frequencies through IL-2 and IL-10 production, respectively, whereas T cells influence stromal cell expression of IL-33 through IL-17A production, and this in turn affects ST2+ Treg numbers in visceral adipose tissue. By producing different sets of cytokines, iNKT and T cells are both able to support adipose Tregs at adolescence and with age, respectively. Note that some studies found that adipose iNKT cells drive proinflammatory, pathogenic immune responses during obesity. These differences have not been reconciled but identifying the mechanisms behind them may reveal further insights into iNKT cell biology and how tissue-specific cues can drive different cell phenotypes [141].
View Figure 15
Figure 15: Upper figure. Cellular changes in adipose tissue in young and aged mice. Young adipose tissue is highly enriched with iNKT cells, which support M2 macrophage and Treg expansion by production of IL-10 and IL-2, respectively. As mice age, iNKT cells are gradually replaced by a population of IL-17A-secreting γδ T cells that support Tregs by a stromal cell-IL-33 axis. Lower figure. Nonredundant actions of iNKT and γδ T cells promote adipose tissue homeostasis. Top panel, Adipose iNKT cells expand Tregs via production of IL-2 and M2 macrophages via production of IL-10. Furthermore, activation of adipose iNKT cells induces weight loss and thermogenesis through an FGF21-dependent mechanism. Bottom panel, IL-17A-producing γδ T cells in adipose tissue drive Treg expansion by inducing stromal cells to produce IL-33. Additionally, a γδ IL-17A axis in adipose tissue is critical for maintenance of body temperature at thermoneutrality and upon cold challenge. iNKT cells and γδ cells, two innate T lymphocytes, have emerged as key regulators of immune homeostasis in visceral adipose tissue, as well as rheostats controlling body temperature in response to environmental fluctuations in sc and brown adipose depots (Figure 2). iNKT cells control adipose Tregs and macrophage frequencies through IL-2 and IL-10 production, respectively, whereas T cells influence stromal cell expression of IL-33 through IL-17A production, and this in turn affects ST2+ Treg numbers in visceral adipose tissue. By producing different sets of cytokines, iNKT and T cells are both able to support adipose Tregs at adolescence and with age, respectively. Note that some studies found that adipose iNKT cells drive proinflammatory, pathogenic immune responses during obesity. These differences have not been reconciled but identifying the mechanisms behind them may reveal further insights into iNKT cell biology and how tissue-specific cues can drive different cell phenotypes [141].
View Figure 15
 Figure 16: Glucose and fatty acids converge to the tricarboxylic acid cycle (TCA), which releases carbons in the form of CO2. Reducing equivalents are also generated in the form of NADH and FADH2 driving the electron transport chain to uncoupled respiration. The physiological importance of metabolic responses to cold exposure or dietary nutrient restriction cannot be understood without first considering the evolutionary pressure for mitochondria to choose fats as a fuel source when systemic glucose reserves are threatened. Fats represent an abundant, carbon rich energy source for many high oxidative tissues. On the contrary, the brain has limited capacity for fat catabolism and therefore relies heavily on a continuous glucose supply. During energy-demanding conditions such as cold exposure, dietary restriction or exercise, glucose catabolism is supported by an increased flux of lipids into adipocytes. This metabolic currency is mediated in large part by noradrenaline, thyroid hormones (triiodothyronine, T3; thyroxine, T4) and glucagon, which exert strong influence on adipocyte lipolysis. In a context of conservative physiology, the metabolic response of activated BAT likely consists in the use of circulating lipids as elective fuel for thermogenesis as a mechanism of protection against low glucose availability. The cycle of feast and famine was an important driving force throughout all human evolution and the adipose tissue has evolved in a context of a continuous coping with dramatic fluctuations in food and nutrient availability. On the contrary, in the modern era human adipose tissue physiology is characterized by an assured daily intake of nutrients. In a pathological view, persistent overnutrition also occurs in adipose tissue and this may lead to a metabolic inflexibility that is characterized by deregulated nutrient and energy homeostasis impairment in resident adipocytes. Therefore, upon chronic overfeeding the mitochondria are metabolically exhausted and trigger an adaptive cell response consisting of the limitation of glucose uptake, i.e., insulin resistance. As stated above, the activity of BAT is beneficial for the prevention of T2D. This was already clear in rodents, wherein activation of BAT by cold exposure or chronic treatment with the β3-adrenergic receptor agonist CL-316243 decreased serum concentrations of glucose and increased peripheral insulin sensitivity in lean mice. In T2D, BAT-mediated glucose clearance could alleviate the demand on pancreatic β-cells for insulin, leading to improvement of β-cell function. β-cell function and insulin sensitivity in peripheral tissues can be improved by thermogenic adipose tissues thanks to their capacity to clear plasma glucose limiting and ectopic lipid deposition. During aging a progressive decline in the mobilization of free fatty acids in thermogenic adipose depots was found, which is accompanied by increased visceral adiposity and failure to maintain core body temperature during cold stress. Adipose tissue macrophages appear to play a key role in age-dependent lipolysis reduction as they lower the bioavailability of noradrenaline. Inflammatory molecules such as IL-1β or TNFα attenuate β adrenergic signalling and cold-induced Ucp1 expression in BAT. A chronic treatment with a low dose of LPS also induces hypothermia by restraining BAT activity via Toll-like receptor 4. Several reports demonstrated that exposure to cool temperature, as well as dietary nutrient restriction, induces skewing toward type 2 immunity and alternative activation of macrophage into the M2 phenotype. Differently to M1 macrophages that express CD11c in addition to CD11b and F4/80 and secrete inflammatory factors including TNF-α, IL-1β, IL-6, leukotriene B4, and nitric oxide M2 macrophages express CD11b, F4/80 and CD206 producing anti -inflammatory cytokines, such as IL-10. Several findings reported that the alternative activation of tissue-resident macrophages is mediated bythe type 2 cytokine IL-4. T-cell-specific Stat6/Pten axis links cold-exposure to Foxp3+Treg induction and adipose tissue function. Cold exposure or β3-adrenergic activation also reduces local inflammation in fat depots by supporting human Treg activity. In cold exposed mice, Foxp3+Tregs were shown to activate BAT-resident M2 macrophages promoting glucose transporter up-regulation together with glycolytic pathway activation. However, adipose tissue-resident macrophages also shown the ability to induce PPARγ-expressing Tregs which are functional to restore inflammatory state of adipose tissue and, hence, insulin sensitivity in obese mice. Obesity results in increased expression of TNF-α in adipose tissue and this promotes insulin resistance has formed the cornerstone for the foundation of tissue immunometabolism. Macrophages represent the predominant immune cells in fat depots and activated inflammatory M1 macrophages have been shown to induce insulin resistance in peripheral tissues such as adipose depots. This metabolic setting leads to increased circulating levels of glucose, which becomes available for infiltrating immune cells to sustain inflammatory inputs. Accordingly, the common feature of insulin resistance states is high glycaemia and low-grade sterile inflammation. A plethora of evidence has reported a higher abundance of M1 inflammatory macrophage infiltrates in the adipose depots of obese than lean subjects. A. Metabolic rewiring and nutrient partitioning between thermogenically-activated adipocytes and distinctly polarized macrophages. Pro-inflammatory M1 macrophages show high glycolytic rate feeding both pentose phosphate pathway for NADPH production and TCA cycle, which however, exhibits two breaks. The first break in TCA occurs at the isocitrate dehydrogenase (IDH) enzyme that results in increased levels of citrate and itaconic acid. High citrate and NADPH levels are the substrates for the synthesis of inflammatory lipids (e.g. prostaglandin). The second break is mediated by itaconate that limits succinate dehydrogenase (SDH) activity thus increasing the intra/extracellular succinate levels. Succinate stabilizes HIF-1α, which binds to the interleukin (IL)-1β promoter, boosting IL-1β production and inflammation. Alternatively-activated M2 macrophages show limited glycolysis. M2 macrophages preferentially catabolize fatty acid through β-oxidation pathway toreplenish TCA resulting in sustained ATP production via OxPHOS activity. Thermogenic adipocytes depend on glucose and circulating fatty acids. A portion of fatty acids is also released from thermogenic adipose cells through lipolysis. The enhancement of glycolysis and fatty acid oxidation serves the uncoupled respiration to produce heat. Thermogenic adipocytes also catabolize high amount of succinate, which is taken up from the circulation and fills TCA cycle enhancing bioenergetics and redox machinery. Our hypothesis is that the ignition of thermogenesis promotes "metabolic hunger" in thermogenic adipocytes channelling a massive number of circulating substrates to uncoupled respiration. The high nutrient flux toward activated adipocytes limits glucose availability for macrophages and increases the circulating fatty acids/glucose ratio, thus forcing M1-to-M2 metabolic rewiring in macrophages (Randle's cycle). Furthermore, the increased succinate uptake by thermogenic adipocytes also limits the succinate uptake in M1macrophages, thus contributing in the slowing-down of the pro-inflammatory features in macrophages. The activation of PPARγ and AMPK skews M2 macrophages and promotes cold-mimicking response in thermogenic adipose tissues. B. Thermogenic adipose tissues and immune-metabolic decline during aging. Aging is accompanied by a physiological loss of thermogenic adipose tissues (brown and subcutaneous), and by a progressive increase of visceral fat. Such adipose tissue remodelling leads to a decrease in circulating thermogenic and anti-inflammatory adipokines (e.g. adiponectin, Neuregulin-4). Sedentary lifestyle and overfeeding during aging may limit the skewing to type 2 immunity of immune cells resident in adipose depots, and favour a shift towards type 1 immunity (increase in M1/M2 ratio) and a setting of a low-grade chronic inflammation. This causes cellular metabolic inflexibility and the development of insulin resistance, which is characterized by high circulating glucose and insulin levels that force the immune metabolism towards an inflammatory phenotype. A systemic disturbance of glucose homeostasis is eventually elicited leading to age-related diseases such as type 2 diabetes and increased vulnerability to immunometabolic injuries (e.g., infections, cancer). Lifestyle interventions, such as physical exercise, intermittent cold exposure, or dietary restrictions, are recommendable preventive strategies: i) To avoid the loss of thermogenic adipose tissues; ii) To maintain protective levels of anti-inflammatory/pro-thermogenic adipokines; iii) To limit the occurrence of low-grade inflammation during aging [132].
View Figure 16
Figure 16: Glucose and fatty acids converge to the tricarboxylic acid cycle (TCA), which releases carbons in the form of CO2. Reducing equivalents are also generated in the form of NADH and FADH2 driving the electron transport chain to uncoupled respiration. The physiological importance of metabolic responses to cold exposure or dietary nutrient restriction cannot be understood without first considering the evolutionary pressure for mitochondria to choose fats as a fuel source when systemic glucose reserves are threatened. Fats represent an abundant, carbon rich energy source for many high oxidative tissues. On the contrary, the brain has limited capacity for fat catabolism and therefore relies heavily on a continuous glucose supply. During energy-demanding conditions such as cold exposure, dietary restriction or exercise, glucose catabolism is supported by an increased flux of lipids into adipocytes. This metabolic currency is mediated in large part by noradrenaline, thyroid hormones (triiodothyronine, T3; thyroxine, T4) and glucagon, which exert strong influence on adipocyte lipolysis. In a context of conservative physiology, the metabolic response of activated BAT likely consists in the use of circulating lipids as elective fuel for thermogenesis as a mechanism of protection against low glucose availability. The cycle of feast and famine was an important driving force throughout all human evolution and the adipose tissue has evolved in a context of a continuous coping with dramatic fluctuations in food and nutrient availability. On the contrary, in the modern era human adipose tissue physiology is characterized by an assured daily intake of nutrients. In a pathological view, persistent overnutrition also occurs in adipose tissue and this may lead to a metabolic inflexibility that is characterized by deregulated nutrient and energy homeostasis impairment in resident adipocytes. Therefore, upon chronic overfeeding the mitochondria are metabolically exhausted and trigger an adaptive cell response consisting of the limitation of glucose uptake, i.e., insulin resistance. As stated above, the activity of BAT is beneficial for the prevention of T2D. This was already clear in rodents, wherein activation of BAT by cold exposure or chronic treatment with the β3-adrenergic receptor agonist CL-316243 decreased serum concentrations of glucose and increased peripheral insulin sensitivity in lean mice. In T2D, BAT-mediated glucose clearance could alleviate the demand on pancreatic β-cells for insulin, leading to improvement of β-cell function. β-cell function and insulin sensitivity in peripheral tissues can be improved by thermogenic adipose tissues thanks to their capacity to clear plasma glucose limiting and ectopic lipid deposition. During aging a progressive decline in the mobilization of free fatty acids in thermogenic adipose depots was found, which is accompanied by increased visceral adiposity and failure to maintain core body temperature during cold stress. Adipose tissue macrophages appear to play a key role in age-dependent lipolysis reduction as they lower the bioavailability of noradrenaline. Inflammatory molecules such as IL-1β or TNFα attenuate β adrenergic signalling and cold-induced Ucp1 expression in BAT. A chronic treatment with a low dose of LPS also induces hypothermia by restraining BAT activity via Toll-like receptor 4. Several reports demonstrated that exposure to cool temperature, as well as dietary nutrient restriction, induces skewing toward type 2 immunity and alternative activation of macrophage into the M2 phenotype. Differently to M1 macrophages that express CD11c in addition to CD11b and F4/80 and secrete inflammatory factors including TNF-α, IL-1β, IL-6, leukotriene B4, and nitric oxide M2 macrophages express CD11b, F4/80 and CD206 producing anti -inflammatory cytokines, such as IL-10. Several findings reported that the alternative activation of tissue-resident macrophages is mediated bythe type 2 cytokine IL-4. T-cell-specific Stat6/Pten axis links cold-exposure to Foxp3+Treg induction and adipose tissue function. Cold exposure or β3-adrenergic activation also reduces local inflammation in fat depots by supporting human Treg activity. In cold exposed mice, Foxp3+Tregs were shown to activate BAT-resident M2 macrophages promoting glucose transporter up-regulation together with glycolytic pathway activation. However, adipose tissue-resident macrophages also shown the ability to induce PPARγ-expressing Tregs which are functional to restore inflammatory state of adipose tissue and, hence, insulin sensitivity in obese mice. Obesity results in increased expression of TNF-α in adipose tissue and this promotes insulin resistance has formed the cornerstone for the foundation of tissue immunometabolism. Macrophages represent the predominant immune cells in fat depots and activated inflammatory M1 macrophages have been shown to induce insulin resistance in peripheral tissues such as adipose depots. This metabolic setting leads to increased circulating levels of glucose, which becomes available for infiltrating immune cells to sustain inflammatory inputs. Accordingly, the common feature of insulin resistance states is high glycaemia and low-grade sterile inflammation. A plethora of evidence has reported a higher abundance of M1 inflammatory macrophage infiltrates in the adipose depots of obese than lean subjects. A. Metabolic rewiring and nutrient partitioning between thermogenically-activated adipocytes and distinctly polarized macrophages. Pro-inflammatory M1 macrophages show high glycolytic rate feeding both pentose phosphate pathway for NADPH production and TCA cycle, which however, exhibits two breaks. The first break in TCA occurs at the isocitrate dehydrogenase (IDH) enzyme that results in increased levels of citrate and itaconic acid. High citrate and NADPH levels are the substrates for the synthesis of inflammatory lipids (e.g. prostaglandin). The second break is mediated by itaconate that limits succinate dehydrogenase (SDH) activity thus increasing the intra/extracellular succinate levels. Succinate stabilizes HIF-1α, which binds to the interleukin (IL)-1β promoter, boosting IL-1β production and inflammation. Alternatively-activated M2 macrophages show limited glycolysis. M2 macrophages preferentially catabolize fatty acid through β-oxidation pathway toreplenish TCA resulting in sustained ATP production via OxPHOS activity. Thermogenic adipocytes depend on glucose and circulating fatty acids. A portion of fatty acids is also released from thermogenic adipose cells through lipolysis. The enhancement of glycolysis and fatty acid oxidation serves the uncoupled respiration to produce heat. Thermogenic adipocytes also catabolize high amount of succinate, which is taken up from the circulation and fills TCA cycle enhancing bioenergetics and redox machinery. Our hypothesis is that the ignition of thermogenesis promotes "metabolic hunger" in thermogenic adipocytes channelling a massive number of circulating substrates to uncoupled respiration. The high nutrient flux toward activated adipocytes limits glucose availability for macrophages and increases the circulating fatty acids/glucose ratio, thus forcing M1-to-M2 metabolic rewiring in macrophages (Randle's cycle). Furthermore, the increased succinate uptake by thermogenic adipocytes also limits the succinate uptake in M1macrophages, thus contributing in the slowing-down of the pro-inflammatory features in macrophages. The activation of PPARγ and AMPK skews M2 macrophages and promotes cold-mimicking response in thermogenic adipose tissues. B. Thermogenic adipose tissues and immune-metabolic decline during aging. Aging is accompanied by a physiological loss of thermogenic adipose tissues (brown and subcutaneous), and by a progressive increase of visceral fat. Such adipose tissue remodelling leads to a decrease in circulating thermogenic and anti-inflammatory adipokines (e.g. adiponectin, Neuregulin-4). Sedentary lifestyle and overfeeding during aging may limit the skewing to type 2 immunity of immune cells resident in adipose depots, and favour a shift towards type 1 immunity (increase in M1/M2 ratio) and a setting of a low-grade chronic inflammation. This causes cellular metabolic inflexibility and the development of insulin resistance, which is characterized by high circulating glucose and insulin levels that force the immune metabolism towards an inflammatory phenotype. A systemic disturbance of glucose homeostasis is eventually elicited leading to age-related diseases such as type 2 diabetes and increased vulnerability to immunometabolic injuries (e.g., infections, cancer). Lifestyle interventions, such as physical exercise, intermittent cold exposure, or dietary restrictions, are recommendable preventive strategies: i) To avoid the loss of thermogenic adipose tissues; ii) To maintain protective levels of anti-inflammatory/pro-thermogenic adipokines; iii) To limit the occurrence of low-grade inflammation during aging [132].
View Figure 16
 Figure 17: Subcellular equilibrium of NAD+. The NAD+ homeostasis is maintained by the biosynthesis, consumption and recycling in differentiate subcellular compartments including the cytosol, the nucleus and the mitochondria. NAD+ precursors including Trp, NA, NR, NMN and NAM are metabolized into NAD+ via Preiss-Handler pathway, de novo pathway and salvage pathway, respectively. NAD+ can receive hydride to yield the reduced form NADH in the metabolic processes including glycolysis, FAO, and the TCA cycle. NADH provides an electron pair to drive the mitochondrial OXPHOS for the generation of ATP and the conversion of lactic acid to pyruvate. The cytosolic and mitochondrial NADH is exchanged through the malate-aspartate shuttle and glycerol-3-phosphate shuttle, while the cytosolic and mitochondrial NADPH is exchanged by the isocitrate-a-KG shuttle. NAD+ can also be phosphorylated into NADP+ by NAD+ kinases including nicotinamide nucleotide transhydrogenase (NNT) and NAD kinases (NADKs). Cytosolic NADP+ is reduced into NADPH by G6PD and 6PGD in the pentose phosphate pathway, and by ME1 in the conversion of malate to pyruvate. Mitochondrial NADPH is produced by IDH2, GLUD, NNT and ME3. The NADPH is required for the activation of NOXs and the synthesis of palmitate. Abbreviation: α-KGDH, alpha-ketoglutarate dehydrogenase; GLUD, glutamate dehydrogenase; NNT, nicotinamide nucleotide transhydrogenase; G3PDH, glyceraldehyde 3-phosphate dehydrogenase; 6PGD, 6-phosphogluconate dehydrogenase; G6PD, glucose-6-phosphate dehydrogenase; GPx, glutathione peroxidases; IDH1/2, isocitrate dehydrogenase 1 and 2; MDH, malate dehydrogenase; ME1/3, malic enzyme; NADK, NAD+ kinase; NOXs, NADPH oxidases; OXPHOS, oxidative phosphorylation; PPP, pentose phosphate pathway; PRx, peroxiredoxin; SDH, succinate dehydrogenase; SOD1-3, superoxide dismutase type 1-3; TCA cycle, tricarboxylic acid cycle; GSH, Glutathione; LDH, Lactate dehydrogenase. From: Xie, N., Zhang, L., Gao, W. et al. NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential.
Figure 17: Subcellular equilibrium of NAD+. The NAD+ homeostasis is maintained by the biosynthesis, consumption and recycling in differentiate subcellular compartments including the cytosol, the nucleus and the mitochondria. NAD+ precursors including Trp, NA, NR, NMN and NAM are metabolized into NAD+ via Preiss-Handler pathway, de novo pathway and salvage pathway, respectively. NAD+ can receive hydride to yield the reduced form NADH in the metabolic processes including glycolysis, FAO, and the TCA cycle. NADH provides an electron pair to drive the mitochondrial OXPHOS for the generation of ATP and the conversion of lactic acid to pyruvate. The cytosolic and mitochondrial NADH is exchanged through the malate-aspartate shuttle and glycerol-3-phosphate shuttle, while the cytosolic and mitochondrial NADPH is exchanged by the isocitrate-a-KG shuttle. NAD+ can also be phosphorylated into NADP+ by NAD+ kinases including nicotinamide nucleotide transhydrogenase (NNT) and NAD kinases (NADKs). Cytosolic NADP+ is reduced into NADPH by G6PD and 6PGD in the pentose phosphate pathway, and by ME1 in the conversion of malate to pyruvate. Mitochondrial NADPH is produced by IDH2, GLUD, NNT and ME3. The NADPH is required for the activation of NOXs and the synthesis of palmitate. Abbreviation: α-KGDH, alpha-ketoglutarate dehydrogenase; GLUD, glutamate dehydrogenase; NNT, nicotinamide nucleotide transhydrogenase; G3PDH, glyceraldehyde 3-phosphate dehydrogenase; 6PGD, 6-phosphogluconate dehydrogenase; G6PD, glucose-6-phosphate dehydrogenase; GPx, glutathione peroxidases; IDH1/2, isocitrate dehydrogenase 1 and 2; MDH, malate dehydrogenase; ME1/3, malic enzyme; NADK, NAD+ kinase; NOXs, NADPH oxidases; OXPHOS, oxidative phosphorylation; PPP, pentose phosphate pathway; PRx, peroxiredoxin; SDH, succinate dehydrogenase; SOD1-3, superoxide dismutase type 1-3; TCA cycle, tricarboxylic acid cycle; GSH, Glutathione; LDH, Lactate dehydrogenase. From: Xie, N., Zhang, L., Gao, W. et al. NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential.
NAD+ metabolism controls the redox homeostasis. ROS could be produced from either metabolic reaction in mitochondria, such as OXPHOS, or from a range of cytosolic enzymes, including NOXs, XO, LOX, CYPs, all of which need the NADH/NADPH serving as the electron donor. To maintain the redox homeostasis, both enzymatic and non-enzymatic antioxidant system components exhibit their effects in coordination with each other to contract with the ROS. GSH, the most abundant of non-enzymatic antioxidants, is synthesized from glutamate, cysteine and glycine catalyzed by two consecutive cytosolic enzymes, GCL and GS. Importantly, NADPH serves as the reductive power for ROS-detoxifying enzymes including glutathione reductases (GR) and thioredoxin reductases (TrxR) to maintain the reduced forms of GSH and Trx (SH)2 in response to ROS produced from mitochondria or NOXs. Abbreviations: 6PGD, 6-phosphogluconate dehydrogenase; CYPs, Cytochromes P450; G6PD, glucose-6-phosphate dehydrogenase; GCL; GR, glutathione reductases; GS; LOX; NAD, nicotinamide adenine dinucleotide; NOXs, NADPH oxidases; NADPH, nicotinamide adenine dinucleotide phosphate; OXPHOS, oxidative phosphorylation; PRx, peroxiredoxin; GPx, glutathione peroxidases; SOD1/2, superoxide dismutase 1 and 2; Trx, thioredoxin; TrxR, thioredoxin reductases; XO, xanthine oxidase [53].
View Figure 17
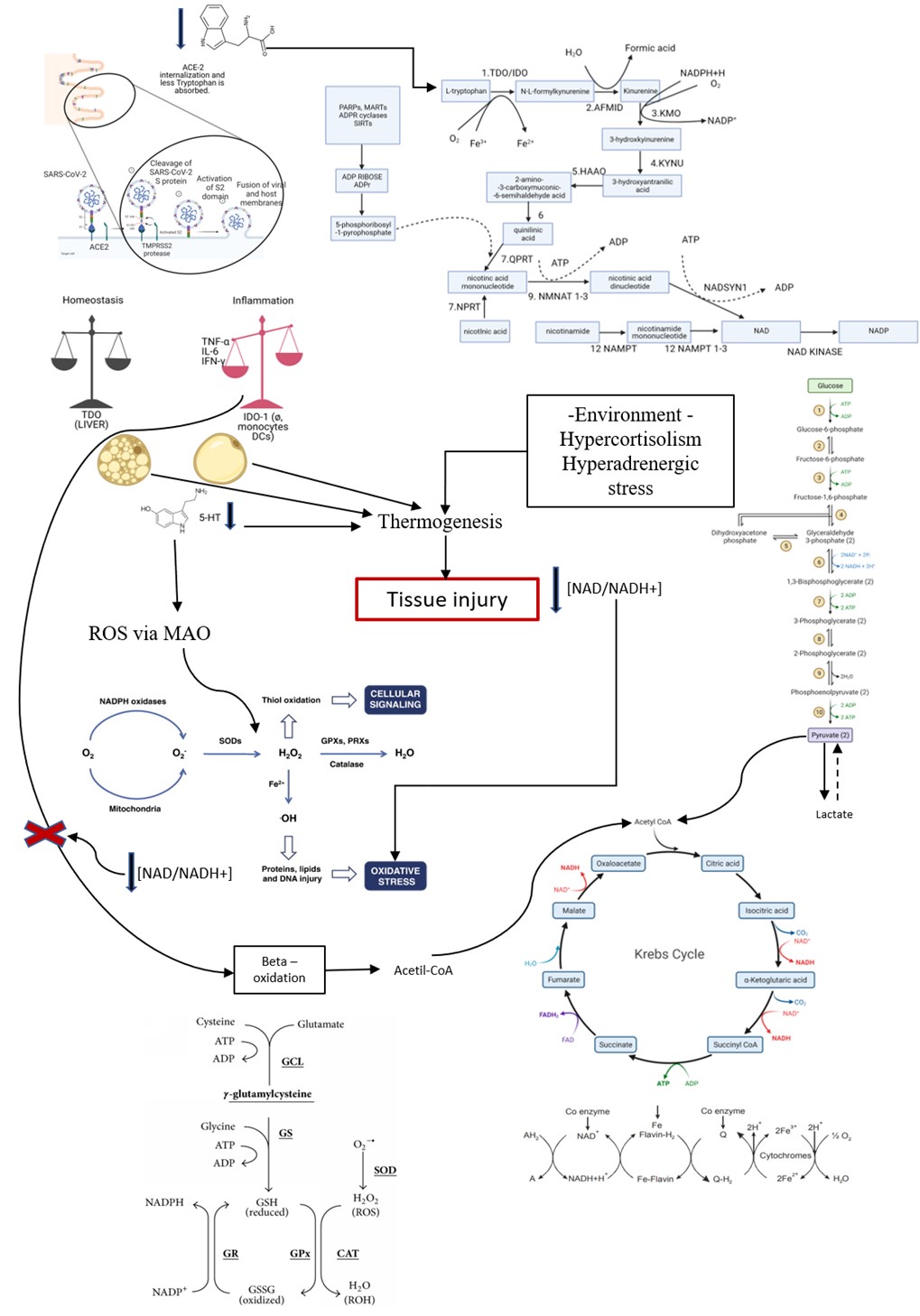 Figure 18: Thermogenesis like Hecatomb. Tryptophan (Trp) is converted into serotonin (5-HT) within the brain by the enzyme tryptophan hydroxylase (TPH)2, where it both increases sympathetic nervous system signalling to brown adipose tissue (BAT) and suppresses appetite. In the gastrointestinal tract (GIT), Trp is converted to serotonin by the enzyme Tph1. Lack of serotonin by inhibition of Tph1 or deleting the Tph1 gene increases BAT cells' sensitivity to noradrenaline (NA) and β-adrenergic receptor (β-AR) signalling. This drives thermogenesis by fat and glucose oxidation via the actions of Ucp1. Sustained thermogenesis recruits additional fat and glucose from peripheral tissues for oxidation by BAT cells, which reduces fat storage in WAT and undesirable locations such as skeletal muscle and the liver, thereby protecting against obesity, skeletal muscle insulin resistance and NAFLD.
Figure 18: Thermogenesis like Hecatomb. Tryptophan (Trp) is converted into serotonin (5-HT) within the brain by the enzyme tryptophan hydroxylase (TPH)2, where it both increases sympathetic nervous system signalling to brown adipose tissue (BAT) and suppresses appetite. In the gastrointestinal tract (GIT), Trp is converted to serotonin by the enzyme Tph1. Lack of serotonin by inhibition of Tph1 or deleting the Tph1 gene increases BAT cells' sensitivity to noradrenaline (NA) and β-adrenergic receptor (β-AR) signalling. This drives thermogenesis by fat and glucose oxidation via the actions of Ucp1. Sustained thermogenesis recruits additional fat and glucose from peripheral tissues for oxidation by BAT cells, which reduces fat storage in WAT and undesirable locations such as skeletal muscle and the liver, thereby protecting against obesity, skeletal muscle insulin resistance and NAFLD.
KP metabolites such as 3-hydroxykynurenine (3HK), 3-hydroxyanthranilic acid (3HAA), and quinolinic acid can significantly inhibit T cell proliferation and induce T cell apoptosis, thereby highlighting the critical roles of other KP metabolites in affecting immune cells. Superoxide is mainly produced by NADPH oxidases and by the mitochondrial respiratory chain. Superoxide is converted to hydrogen peroxide (H2O2) by superoxide dismutases (SODs). H2O2 is converted to water (H2O) by glutathione peroxidases (GPX), peroxiredoxins (PRX) or catalase. H2O2 is the main player in ROS cellular signalling because it can promote posttranslational modifications in proteins by thiol oxidation. The reaction of H2O2 with iron (Fe2+) generates hydroxyl radicals (radical dotOH) responsible for lipid, protein and DNA damage, promoting oxidative stress.
Brown adipose tissue (BAT), with its abundance of UCP1 positive mitochondria, was thought to be found in distinct adipose tissue depots, separate from the more abundant WAT found subcutaneously and in the viscera. BAT could be induced within adipose tissue depots typically considered as WAT. Treatment with a β3-adrenoreceptor agonist also induced UCP1 expression within WAT, implicating adrenergic stress in this process. In COVID-19, severity in obese and diabetic patients is magnified by the stressor environment responsible for the hyperadrenergic stimuli. When 5-HT is degraded to ROS formation via MAO, the microenvironment is already lacking niacin (NAD/NADH+), and the fats also deviate from the beta-oxidation pathway to the ROS formation pathway. Adipocytes, whether brown or white, are mobilized for a catastrophic thermogenic pathway that will cause generalized tissue damage and be responsible for maintaining the inflammatory process at the expense of minimal change in the environment. In other words, small stressors serve as fuel for the maintenance of inflammation in COVID-19. Figure 1 also shows the glutamine/glutathione pathway, whose purpose is to show the importance of N-acetylcysteine (Nac) as a drug to be provided for these severe cases. Acetylcysteine is a derivative of the natural amino acid cysteine, which serves as a substrate for the synthesis of glutathione (GSH) in the body, which has an antioxidant effect. This reduces the formation of pro-inflammatory cytokines, such as IL-9 and TNF-α.
The scale shows that during an inflammatory process, the metabolism of tryptophan ceases to be carried out, mostly by hepatic TDO and starts to be metabolized by IDO-1 produced by macrophages, dendritic cells, and monocytes mainly. This pathway promotes immunotolerance as the primary response of immune cells to this alteration of metabolism amid inflammation.
View Figure 18
The metabolism of the amino acids Try, Phe and Tyr, support the hypothesis that low-grade inflammation is responsible for significant correlations found between immune system biomarkers like neopterin, KYN /Try or Phe/Tyr. A link between increased KYN levels and an elevated KYN/Try is associated with accelerated Try breakdown due to activation of the enzyme IDO. Deprivation of TRP is an efficient strategy during the Th1-type immune response to counteract the unwanted proliferation of pathogens, infected cells and tumour cells. Immune activation and Phe/Tyr results from the disturbed activity of Phe-metabolizing enzyme PAH due to insufficient supply with its cofactor BH4, which is sensitive to oxidation.
Kynurenine - a tryptophan metabolite that blocks antitumour immunity via Treg activation and Teff apoptosis - inhibited T cell proliferation in a manner that BH4 can rescue. Accumulation of neuroactive kynurenine metabolites, such as quinolinic acid, may contribute to neurological/psychiatric disorders [61,150,151].
The biomarker most associated with tryptophan metabolites is neopterin, which had positive associations with KTR, kynurenine and anthranilic acid. Elevated levels of neopterin are associated with a pro-inflammatory state mediated by activation of the cellular immune system. It is an endogenous regulator of cytotoxic effects by activated macrophages and a potent peroxynitrite enhancer. It is a stable molecule eliminated only by the kidney. Higher levels are associated with higher reactive oxygen species and serve as an estimate of oxidative stress caused by the immune system. Th1-type IFN-γ and tumour necrosis factor-α may lead to an increase in neopterin, which is a biomarker of activated cell-mediated immunity. Neopterin was also negatively associated with tryptophan [152].
IL-10 is the most associated immune system biomarker, positively associated with kynurenine, KTR, 3-hydroxykynurenine and 3-hydroxyanthranilic acid. TNFα is negatively associated with tryptophan. Monocyte chemoattractant protein 1 is positively associated with tryptophan and, IFNγ is not associated with tryptophan metabolites. CD25+ Foxp3+ regulatory T lymphocytes are potent inducers of IL-10; this mechanism would allow kynurenine to exert a negative feedback loop on an initial acute inflammatory event and maintain a continuous balance between inflammation and immunosuppressive activity [28,49,54,73,132,133,153,154].
(6R)-5,6,7,8-tetrahydrobiopterin (BH4) is an obligate cofactor of nitric oxide (NO) synthases, playing a redox role in the catalysis of NO formation from L-arginine, O2 and NADPH. BH4 is also a required cofactor for the phenylalanine, tyrosine and tryptophan hydroxylases. The biosynthesis of BH4 is dependent on the activity of the rate-limiting enzyme GTP-cyclohydrolase I (GTPCH I). EPO causes an increase in intracellular levels of BH4 via activation of GTPCH I. Inhibition of Jak2 abolishes EPO-induced BH4 biosynthesis, suggesting that increased phosphorylation and activation of Jak2 are the molecular mechanisms underlying the observed effect of EPO on BH4 synthesis. PI3K activity is an upstream activator of Akt1 because pharmacological and genetic inactivation of PI3K/Akt1 abolishes the stimulatory effects of EPO on GTPCH I activity and biosynthesis of BH4 in mouse aorta.
IL-1β, interferon-γ and TNF-α, can induce both GCH1 expression and activity, therefore increasing BH4 synthesis. At the post-translational level, GCH1 is inhibited by BH4 and stimulated by phenylalanine [153-156].
Inflammatory stimulation activates the inducible NOS, which increases the use of BH4 for optimal enzymatic activity and induces the formation of large amounts of oxygen radicals that contribute to the oxidative loss of BH4. Both increased use and loss of BH4 driven by a chronic inflammatory state may synergistically alter the function of BH4-dependent enzymes and then compromise the biosynthesis of monoamines, which may contribute to mood disorders. Thus, increased xanthurenic acid, a metabolite of 3-HK, has been shown to directly lower BH4 biosynthesis by inhibiting sepiapterin reductase. Concurrent upregulation of kynurenines and BH2 production may lead to a combined up-regulated activity of NOS (by kynurenines) and decreased availability of BH4. Such a combination results in an uncoupling of NOS and consequently reduced NO production favouring reactive oxygen species (ROS), which cause further oxidative BH4 loss.
Genetic inactivation of GTP cyclohydrolase 1 (GCH1, the rate-limiting enzyme in the synthesis of BH4) and inhibition of sepiapterin reductase (the terminal enzyme in the synthetic pathway for BH4) severely impair the proliferation of mature mouse and human T cells. BH4 production in activated T cells is linked to alterations in iron metabolism, mitochondrial bioenergetics and blockade of BH4 synthesis abrogate T-cell-mediated autoimmunity and allergic inflammation and enhancing BH4 levels through GCH1 overexpression augments responses by CD4+ and CD8+ expressing T cells, increasing their antitumour activity in vivo [8,13,61,157].
These inflammatory/anti-inflammatory pathways are related to the development of mood disorders and neurological diseases. Disruption of monoamine metabolism leads to several neurological manifestations in childhood that are evident in the clinical history and physical examination. The presentation of disorders can include cognitive and motor delay, epilepsy, autonomic dysfunction (which manifests as sweating, temperature dysregulation, hypersalivation and nasal congestion) and neuropsychiatric features such as anxiety or autism spectrum disorder. Motor symptoms are often prominent and include gait disturbances, dystonia, dyskinesia, parkinsonism, tremor, oculogyric crises, eyelid ptosis, and axial hypotonia; motor symptoms become more prominent at night and improve after sleep. Other associated features include feeding difficulties and microcephaly. Many clinical features of monoamine neurotransmitter disorders are seen in other neurological conditions, such as cerebral palsy, primary movement disorders, paroxysmal disorders, hypoxic-ischemic encephalopathy, and epileptic encephalopathies (Figure 19, Figure 20, Figure 21 and Figure 22).
 Figure 19: Homocysteine is an amino acid derivative from the metabolism of the sulphur-containing amino acid methionine, which is present in proteins found in meats, poultry, dairy products, eggs, and fish. Methionine is converted into S-adenosylmethionine (SAMe), which participates in numerous one-carbon methylation (CH3) reactions in the body, including those that create an essential phospholipid (phosphatidylcholine) and neurotransmitters (serotonin, melatonin, epinephrine, dopamine). After donation of its methyl group, SAMe becomes S-adenosylhomocysteine, then homocysteine. At this point, homocysteine must either be further metabolized via transculturation to become cysteine, taurine, and glutathione - a B6-dependent process - or re-methylated to become methionine again. Re-methylation is done via one of two reactions: methionine synthetase facilitates the donation of a methyl group from methyl cobalamin (vitamin B12, which gets its methyl group from 5-MTHFor betaine-homocysteine methyl-transferase facilitates donation of a methyl group from betaine (trimethyl glycine). Epidemiologically, low blood levels of folate and vitamin B12 and high levels of homocysteine have been correlated with depression, especially in the elderly. Folate appears to be important in regenerating BH4, which is highly susceptible to oxidation. The folate-metabolizing enzyme dihydrofolate reductase might also be involved in BH4 regeneration.36 Other research suggests folate is necessary as a starting material for pterin synthesis and this may be the focus of the folate/BH4 relationship. Folate and BH4 share a second biochemical pathway. In vascular endothelial cells, endothelial nitric oxide synthase (eNOS) is the enzyme responsible for creating nitric oxide. BH4 is the nutrient cofactor for this enzyme. It has been demonstrated that folate, in the form of 5-MTHF, regenerates oxidized BH4,37 and in the absence of an adequate amount of BH4, 5-MTHF "stands in" for BH4 at the enzyme level.38 The chemical structures of BH4 and 5-MTHF are similar enough that eNOS will accept 5-MTHF as a substitute cofactor (Figure 5). 39 A similar mechanism might be at play in the antidepressant effect of folate. The Methylation, Neurotransmitter, and Antioxidant Connections Between Folate and Depression. Available from: BH4 = tetrahydrobiopterin; 5-MTHF = 5-methyltetrathydrofolate; H-hydrogen; CH3 = methyl group; CH2 = methylene; THF = tetrahydrofolate; MTHFR = methylene tetrahydrofolate reductase; qBH = quinonoid dihydropterin; NADH-nicotinamide adenine dinucleotide; NADP = nicotinamide adenine dinucleotide phosphate; NAD = nicotinamide adenine dinucleotide (oxidized); NADPH = nicotinamide adenine dinucleotide phosphate (reduced); DHF = dihydrofolate; DHFR = dihydrofolate reductase; GTP = guanosine triphosphate [171].
View Figure 19
Figure 19: Homocysteine is an amino acid derivative from the metabolism of the sulphur-containing amino acid methionine, which is present in proteins found in meats, poultry, dairy products, eggs, and fish. Methionine is converted into S-adenosylmethionine (SAMe), which participates in numerous one-carbon methylation (CH3) reactions in the body, including those that create an essential phospholipid (phosphatidylcholine) and neurotransmitters (serotonin, melatonin, epinephrine, dopamine). After donation of its methyl group, SAMe becomes S-adenosylhomocysteine, then homocysteine. At this point, homocysteine must either be further metabolized via transculturation to become cysteine, taurine, and glutathione - a B6-dependent process - or re-methylated to become methionine again. Re-methylation is done via one of two reactions: methionine synthetase facilitates the donation of a methyl group from methyl cobalamin (vitamin B12, which gets its methyl group from 5-MTHFor betaine-homocysteine methyl-transferase facilitates donation of a methyl group from betaine (trimethyl glycine). Epidemiologically, low blood levels of folate and vitamin B12 and high levels of homocysteine have been correlated with depression, especially in the elderly. Folate appears to be important in regenerating BH4, which is highly susceptible to oxidation. The folate-metabolizing enzyme dihydrofolate reductase might also be involved in BH4 regeneration.36 Other research suggests folate is necessary as a starting material for pterin synthesis and this may be the focus of the folate/BH4 relationship. Folate and BH4 share a second biochemical pathway. In vascular endothelial cells, endothelial nitric oxide synthase (eNOS) is the enzyme responsible for creating nitric oxide. BH4 is the nutrient cofactor for this enzyme. It has been demonstrated that folate, in the form of 5-MTHF, regenerates oxidized BH4,37 and in the absence of an adequate amount of BH4, 5-MTHF "stands in" for BH4 at the enzyme level.38 The chemical structures of BH4 and 5-MTHF are similar enough that eNOS will accept 5-MTHF as a substitute cofactor (Figure 5). 39 A similar mechanism might be at play in the antidepressant effect of folate. The Methylation, Neurotransmitter, and Antioxidant Connections Between Folate and Depression. Available from: BH4 = tetrahydrobiopterin; 5-MTHF = 5-methyltetrathydrofolate; H-hydrogen; CH3 = methyl group; CH2 = methylene; THF = tetrahydrofolate; MTHFR = methylene tetrahydrofolate reductase; qBH = quinonoid dihydropterin; NADH-nicotinamide adenine dinucleotide; NADP = nicotinamide adenine dinucleotide phosphate; NAD = nicotinamide adenine dinucleotide (oxidized); NADPH = nicotinamide adenine dinucleotide phosphate (reduced); DHF = dihydrofolate; DHFR = dihydrofolate reductase; GTP = guanosine triphosphate [171].
View Figure 19
 Figure 20: Schematic sketch of the interrelationships between some of the most relevant reactive oxygen species (ROS) that affect the vascular wall. Superoxide (O2-∙) is produced from molecular oxygen (O2) by different sources, such as nicotinamide adenine dinucleotide phosphate - NADPH oxidase (NOx), mitochondrial respiratory chain, xanthine oxidase, uncoupled endothelial nitric oxide synthase (eNOS) and lipoxygenases. Superoxide can directly affect vascular cells, but it can also be converted by superoxide dismutases (SOD) to hydrogen peroxide (H2O2). H2O2 can undergo spontaneous conversion to a hydroxyl radical (OH∙, extremely reactive - it attacks most cellular components) in the presence of iron (Fe2+) through the Fenton reaction. H2O2 produces direct effects on the vascular wall or can be detoxified via glutathione peroxidase (GPx), catalase (Cat) or thioredoxin (Trx) peroxidase for H2O and O2. Superoxide can also react with nitric oxide (NO) or arachidonic acid to form peroxynitrite (ONOO⋅-) or isoprostanes, respectively. In addition to other signaling effects, H2O2 can activate Nox, resulting in increased superoxide production. The enzyme myeloperoxidase (MPO) can use H2O2 to oxidize the chloride to the strong oxidizing agent hypochlorous acid (HOCl). HOCl can chlorinate and thus inactivate several biomolecules, including lipoproteins and the eNOS substrate L-arginine. In addition to generating HOCl, myeloperoxidase can oxidize (and thus inactivate) NO to nitrite (NO2-) in the vasculature. Paraoxonase (PON) isoforms 2 and 3 can prevent mitochondrial generation of O2. Uncoupled eNOS generates O∙-2 instead of NO due to low levels of its cofactor tetrahydrobiopterin (BH4) or its substrate L-arginine (Alp and Channon, 2004). ROS, in particular peroxynitrite, promotes "uncoupling" eNOS (Sena et al., 2013; Santilli et al., 2015). Superoxide reacts with NO forming peroxynitrite that further oxidizes BH4 to dihydrobiopterin (BH2), creating a vicious circle and more eNOS uncoupling (Li and Forstermann, 2014). Under physiological conditions, PVAT prevents eNOS uncoupling [172].
View Figure 20
Figure 20: Schematic sketch of the interrelationships between some of the most relevant reactive oxygen species (ROS) that affect the vascular wall. Superoxide (O2-∙) is produced from molecular oxygen (O2) by different sources, such as nicotinamide adenine dinucleotide phosphate - NADPH oxidase (NOx), mitochondrial respiratory chain, xanthine oxidase, uncoupled endothelial nitric oxide synthase (eNOS) and lipoxygenases. Superoxide can directly affect vascular cells, but it can also be converted by superoxide dismutases (SOD) to hydrogen peroxide (H2O2). H2O2 can undergo spontaneous conversion to a hydroxyl radical (OH∙, extremely reactive - it attacks most cellular components) in the presence of iron (Fe2+) through the Fenton reaction. H2O2 produces direct effects on the vascular wall or can be detoxified via glutathione peroxidase (GPx), catalase (Cat) or thioredoxin (Trx) peroxidase for H2O and O2. Superoxide can also react with nitric oxide (NO) or arachidonic acid to form peroxynitrite (ONOO⋅-) or isoprostanes, respectively. In addition to other signaling effects, H2O2 can activate Nox, resulting in increased superoxide production. The enzyme myeloperoxidase (MPO) can use H2O2 to oxidize the chloride to the strong oxidizing agent hypochlorous acid (HOCl). HOCl can chlorinate and thus inactivate several biomolecules, including lipoproteins and the eNOS substrate L-arginine. In addition to generating HOCl, myeloperoxidase can oxidize (and thus inactivate) NO to nitrite (NO2-) in the vasculature. Paraoxonase (PON) isoforms 2 and 3 can prevent mitochondrial generation of O2. Uncoupled eNOS generates O∙-2 instead of NO due to low levels of its cofactor tetrahydrobiopterin (BH4) or its substrate L-arginine (Alp and Channon, 2004). ROS, in particular peroxynitrite, promotes "uncoupling" eNOS (Sena et al., 2013; Santilli et al., 2015). Superoxide reacts with NO forming peroxynitrite that further oxidizes BH4 to dihydrobiopterin (BH2), creating a vicious circle and more eNOS uncoupling (Li and Forstermann, 2014). Under physiological conditions, PVAT prevents eNOS uncoupling [172].
View Figure 20
 Figure 21: In vascular disease states such as atherosclerosis and diabetes, endothelial nitric oxide (NO) bioactivity is diminished and oxidative stress is increased, resulting in endothelial dysfunction. It has become apparent that enzymatic "coupling" of endothelial NO synthase (eNOS) by the cofactor tetrahydrobiopterin (BH4) plays a key role in maintaining endothelial function. Indeed, the balance between NO and superoxide production by eNOS seems to be determined by the availability of BH4, whereas BH4 oxidation forms 7,8-dihydrobiopterin (BH2), which is inactive for NOS cofactor function and may compete with BH4 for NOS binding and increase eNOS uncoupling. Th1-type cytokine IFN-γ was elucidated as the primary driving force of neopterin production in human and primate monocyte-derived cells like monocytes-macrophages, dendritic cells, and in specific astrocyte-derived cells. However, other proinflammatory stimuli like IFN-α and -β or LPS can also induce neopterin formation, although to a lesser extent. In human monocyte cells, increased NO. Production is hampered even after stimulation with IFN-γ because neopterin and 7,8-dihydroneopterin are produced at the expense of BH4. Other human cell types can generate BH4 and thus forming NO. once iNOS is activated [25]. The presence of this biosynthetic pathway enables both cells to intracellularly produce L-tyrosine from the essential amino acid L-phenylalanine via phenylalanine hydroxylase (PAH) (EC 1.14.16.1) in the presence of oxygen. L-tyrosine serves as the substrate for tyrosine hydroxylase (EC 1.14.16.2), together with 6BH4 to produce catecholamines in keratinocytes. In melanocytes, this amino acid is the substrate for tyrosinase, the key enzyme for melanogenesis. Only recently, a new function for 6BH4 was recognized by controlling the initial pigmentation step in epidermal melanocytes. The disturbed tryptophan-serotonin pathway is not the only biochemical route relevant to the development of neuropsychiatric symptoms, e.g., the phenylalanine-tyrosine-dopamine pathway and corresponding biogenic amines are of comparable significance. Another set of critical factors is represented by kynurenine metabolites that act as agonists (quinolinic acid) or antagonists (kynurenic acid) of the N-methyl D-aspartate (NMDA) receptor.
Figure 21: In vascular disease states such as atherosclerosis and diabetes, endothelial nitric oxide (NO) bioactivity is diminished and oxidative stress is increased, resulting in endothelial dysfunction. It has become apparent that enzymatic "coupling" of endothelial NO synthase (eNOS) by the cofactor tetrahydrobiopterin (BH4) plays a key role in maintaining endothelial function. Indeed, the balance between NO and superoxide production by eNOS seems to be determined by the availability of BH4, whereas BH4 oxidation forms 7,8-dihydrobiopterin (BH2), which is inactive for NOS cofactor function and may compete with BH4 for NOS binding and increase eNOS uncoupling. Th1-type cytokine IFN-γ was elucidated as the primary driving force of neopterin production in human and primate monocyte-derived cells like monocytes-macrophages, dendritic cells, and in specific astrocyte-derived cells. However, other proinflammatory stimuli like IFN-α and -β or LPS can also induce neopterin formation, although to a lesser extent. In human monocyte cells, increased NO. Production is hampered even after stimulation with IFN-γ because neopterin and 7,8-dihydroneopterin are produced at the expense of BH4. Other human cell types can generate BH4 and thus forming NO. once iNOS is activated [25]. The presence of this biosynthetic pathway enables both cells to intracellularly produce L-tyrosine from the essential amino acid L-phenylalanine via phenylalanine hydroxylase (PAH) (EC 1.14.16.1) in the presence of oxygen. L-tyrosine serves as the substrate for tyrosine hydroxylase (EC 1.14.16.2), together with 6BH4 to produce catecholamines in keratinocytes. In melanocytes, this amino acid is the substrate for tyrosinase, the key enzyme for melanogenesis. Only recently, a new function for 6BH4 was recognized by controlling the initial pigmentation step in epidermal melanocytes. The disturbed tryptophan-serotonin pathway is not the only biochemical route relevant to the development of neuropsychiatric symptoms, e.g., the phenylalanine-tyrosine-dopamine pathway and corresponding biogenic amines are of comparable significance. Another set of critical factors is represented by kynurenine metabolites that act as agonists (quinolinic acid) or antagonists (kynurenic acid) of the N-methyl D-aspartate (NMDA) receptor.
Phenylketonuria (PKU) is the most prevalent disorder caused by an inborn error in amino acid metabolism, but it is curable. The prevalence of it varies widely around the world (1). PKU is characterized by (Phe) accumulation mainly due to hepatic phenylalanine hydroxylase (PAH) deficiency, which converts Phe to tyrosine (Tyr), requiring the cofactor tetrahydrobiopterin (BH4), molecular oxygen and iron. BH4 is the essential cofactor for PAH and the metabolism of catecholamines, serotonin, and nitric oxide in the central nervous system (CNS). In rare cases, the first sign of PKU develops in late adulthood, resembling common manifestations of neurological diseases such as progressive dementia, spastic paraplegia, ataxia, tremor, and behavioural problems. In blood, Phe competes with other LNAAs, including tryptophan (Try) and Tyr, for L-amino acid transporter 1 (LAT1), which is responsible for their uptake into the brain (1). Thus, HPA simultaneously increases brain Phe levels and reduces other amino acid levels, affecting neurotransmitter production, protein synthesis, myelin metabolism, and glutamatergic synaptic transmission, resulting in impairment in neuropsychological function. Fogginess, concentration reduction, low attention, anxiety, irritability, memory deficit, headache, and unstable mood were standard features in patients with uncontrolled blood phenylalanine levels (generally above 600 µmol/L).
The proinflammatory cytokine IFN-γ is preferentially released during the process of Th1-type immune activation. The cytokine induces the expression of the enzyme GCH, which, in most human cells and cells from other species, results in cofactor BH4. Only human and primate monocyte-derived macrophages and dendritic cells produce neopterin instead of BH4. The enzyme PAH converts Phe to Tyr in the liver and kidneys, requiring BH 4 as a cofactor. Tyr deficiency may cause an insufficient production of biogenic amine neurotransmitters noradrenaline (norepinephrine) and adrenaline (epinephrine), which can underlie the development of mood changes and fatigue in patients with inflammatory conditions. Significant correlations between Phe metabolism and concentrations of immune activation marker neopterin were reported from patients after trauma, with ovarian cancer, with HIV-1 infection, with coronary artery disease, as well as in the healthy elderly, and neopterin concentrations followed the course of other immune activation markers such as interleukin 6, soluble interleukin 2 receptor α, and the 75-kDa TNF receptor. In patients with psychiatric disorders including major depression and schizophrenia, BH 4deficiency was described to relate to the development of neuropsychiatric symptoms. Results confirm the assumption that the impaired conversion of Phe to Tyr in the liver and the concomitantly decreased Tyr availability affect the transport of the amino acid into the brain and its further conversion to its downstream metabolites L-DOPA, dopamine, epinephrine, and norepinephrine. Moreover, A (IgA) and immunoglobulin G (IgG) concentrations in PKU children were significantly decreased as compared to values from similar aged healthy children, a finding that could be secondary to a poor nutritional status of Phe-restrict diet therapy. Calcium homeostasis is crucial for brain function and its dysregulation in PKU was suggested by several works. In this context, parathyroid hormone (hormone that regulates calcium metabolism), osteocalcin, and dihydroxycholecalciferol were found increased in serum samples from phenylketonuric infants, but calcitonin was found reduced. These alterations were not reverted by Phe-restricted diet.
View Figure 21
 Figure 22: Primary disorders of dopamine and serotonin metabolism are attributable to enzyme or cofactor deficiencies, defective neurotransmitter transport and/or reuptake or defective vesicle formation and/or packaging. Neurotransmitter abnormalities are becoming increasingly recognized as secondary phenomena that result from other neurological disorders. Abbreviations: BH4, tetrahydrobiopterin; DAT, dopamine transporter; GTP-CH 1, GTP cyclohydrolase 1; PITX3, pituitary homeobox 3; PTPS, 6-pyruvoyl tetrahydropterin; VMAT2, vesicular monoamine transporter 2 [145].
View Figure 22
Figure 22: Primary disorders of dopamine and serotonin metabolism are attributable to enzyme or cofactor deficiencies, defective neurotransmitter transport and/or reuptake or defective vesicle formation and/or packaging. Neurotransmitter abnormalities are becoming increasingly recognized as secondary phenomena that result from other neurological disorders. Abbreviations: BH4, tetrahydrobiopterin; DAT, dopamine transporter; GTP-CH 1, GTP cyclohydrolase 1; PITX3, pituitary homeobox 3; PTPS, 6-pyruvoyl tetrahydropterin; VMAT2, vesicular monoamine transporter 2 [145].
View Figure 22
Mechanism of action is inhibition of HMG-CoA reductase, the rate-limiting enzyme of cholesterol synthesis. Inhibition of this enzyme interferes with the synthesis of mevalonate and isoprenoid intermediates, particularly farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP), which are responsible for the isoprenylation of a wide range of proteins including small proteins associated with guanosine triphosphate (GTP) such as Ras, Rho and Rac. Isoprenylation of these molecules is essential for the covalent attachment, subcellular localization, and intracellular trafficking of membrane-associated proteins. Inhibiting the synthesis of isoprenoid intermediates may underlie many statins' pleiotropic effects, including their antioxidant and anti-inflammatory action. Statins also manifest immune-modulatory effects by activating regulatory T cells (Tregs). For example, in a murine model of tumour growth, statins activate Treg and increase the production of the immune regulatory markers IL-10 and TGF-β1. Statins reduce the number and increase the suppressive function of Treg cells in experimental animal models of chronic immune activation and humans with or without immune-mediated conditions. The production of Tregs is linked with the production of reactive oxygen species (ROS) geared at a minimum toward eliminating pathogens, at most, to regulate the balance between proinflammatory and regulatory arms of the immune response. This has a narrow regulatory window as specifically unopposed ROS production can suppress regulatory T cell production favouring proinflammatory reactions, or tiny ROS production may also impair Tregs' differentiation, stability, and suppressive function.
Statins reduce the production of kynurenine and actively limit the effects of kynurenine's main excitotoxic metabolite, QA. For example, in a model of QA-induced neurotoxicity in rats, statins significantly decrease the excitotoxic effect of QA, levels of markers of oxidative stress, and proinflammatory cytokines (such as TNF-α) as well as striatal lesion volume. In another study, statins appear to have a neuroprotective effect in excitotoxic rodent seizure models. Specifically, atorvastatin significantly reduced QA-induced clonic or tonic seizures and prevented cell death induced by QA in the hippocampus. Atorvastatin also counteracts the decrease in glutamate uptake triggered by QA and prevents the QA-induced decrease in protein kinase B (PKB, or Akt) phosphorylation.
Lipophilic statins included prescriptions for simvastatin, atorvastatin, pitavastatin, and lovastatin. The hydrophilic statins included rosuvastatin, pravastatin, and Fluvastatin.
|
Recommendation To prescribe: Atorvastatin 80 mg/day |
I assume that we are in a pandemic, so mathematically, those who have not had COVID-19 will still have it, and those who have, unfortunately, can have it again.
We are used to facing diseases with a similar pattern regarding the production of immunoglobulins (IgM and IgG), but it is strange to observe, for example, a family with 5 people sharing the same house. One of them is infected. All other people had contact with the index case, but 2 were asymptomatic, one was oligosymptomatic, and the other had to be hospitalized.
After 14 days, the formation of antibodies is bizarre. Sometimes the one who presented the formation of antibodies was one of the asymptomatic family members.
We must not forget that 60% of cases are asymptomatic or go unnoticed by the patient.
As we have seen, SARS-CoV-2 causes changes in the immune response from its entry into the cell, acting on cathepsins and furins, shifting the tryptophan axis to the Kynurenine pathway. Severe cases have presented intense lymphopenia with apoptosis of CD4+, CD8+, NK (natural killer) and B cells. This apoptosis varies from person to person due to its comorbidities and genetic variations. For example, some studies demonstrate that women have a higher prevalence in forming antibodies to COVID-19, relating to the fact that the innate immunity gene is contained in the X chromosome. Men have a single X; women have two X chromosomes, so they need to be silenced. The pattern of "silencing" occurs in such a way as to form a mosaic, with the woman having functioning X chromosomes of both maternal and paternal origin. This fact seems to promote more significant variability in the identification of pathogens and antibody production.
What seems to happen is that we produce antibodies, but often at low titers that are not identified by the usual tests. That would be the explanation for such a good response to the first dose of the vaccine against SARS-CoV-2. In other words, the first dose was a booster for many people who had COVID-19 but with negative serology.
Another important fact is that most of the cases admitted to the hospital ICU where I work had two doses of the vaccine, which did not prevent them from developing the severe disease.
We must remember that IgG is suitable when it is formed in sufficient quantity to neutralize the virus. Often, little IgG production can cause the phenomenon of infection amplification by the Antibody-dependent enhancement mechanism.
Once I understand that the virus causes immunosuppression, we must re-evaluate the issue of vaccination in patients who have severe disease. Will this patient respond to the vaccine in a good way, or could it be that they will have significant side effects, thinking that the pathophysiology of the disease can promote the development of autoimmune diseases that can be reinforced with vaccination?
I believe that, above all, people's safety must prevail. Producing vaccines without knowing how the virus works may not be appropriate. I am not against vaccines, but I am in favour of people undergoing safe and effective vaccination.
The use of corticosteroids is not controversial when used at the correct time. In the acute phase of the disease, the best period to be used is in the defervescence phase (between D6 and D8 with CPR < 10), avoiding the neutrophilic march that will start the cytokine storm. Corticosteroid pulse at this stage blocks disease progression in patients with predictors of severity and orotracheal intubation is avoided (Figure 23).
 Figure 23: The rationale behind corticosteroid therapy. Upper figure. COVID-19 progression concerning viral kinetics, week of infection. A. the patient who evolves with a leukemoid reaction and neutrophilia is sometimes mistakenly diagnosed as acute leukaemia, but it is common in Sars-CoV-2 infection in patients who evolve severely. B. indicates patients who develop neutropenia and who, in general, are discharged after 14 days of illness. Lower figure. Immunosuppression initiated with the cytokine storm followed by the formation of immunological heterogeneity responsible for the controversy regarding the use of corticosteroids. S1: triangle's area represents patients with lesser and heterogenous immunosuppression; S2: triangle's area allocates patients with more immunosuppression and heterogeneity. Thus, pulse therapy is indicated before the immunosuppressive phase, initiated with the peak of reactive Protein C. The initial phase shows some inflammatory comorbidities, MHC class II genes and chronic infections responsible for the chronic inflammatory profile of patients who progress to severe forms of the disease. It is necessary to emphasize that even lymphocyte recovery is not enough to restore normal immunity, since many articles already show that the recovered lymphocytes, in great majority, are in exhaustion expressing PD-1 protein [68].
View Figure 23
Figure 23: The rationale behind corticosteroid therapy. Upper figure. COVID-19 progression concerning viral kinetics, week of infection. A. the patient who evolves with a leukemoid reaction and neutrophilia is sometimes mistakenly diagnosed as acute leukaemia, but it is common in Sars-CoV-2 infection in patients who evolve severely. B. indicates patients who develop neutropenia and who, in general, are discharged after 14 days of illness. Lower figure. Immunosuppression initiated with the cytokine storm followed by the formation of immunological heterogeneity responsible for the controversy regarding the use of corticosteroids. S1: triangle's area represents patients with lesser and heterogenous immunosuppression; S2: triangle's area allocates patients with more immunosuppression and heterogeneity. Thus, pulse therapy is indicated before the immunosuppressive phase, initiated with the peak of reactive Protein C. The initial phase shows some inflammatory comorbidities, MHC class II genes and chronic infections responsible for the chronic inflammatory profile of patients who progress to severe forms of the disease. It is necessary to emphasize that even lymphocyte recovery is not enough to restore normal immunity, since many articles already show that the recovered lymphocytes, in great majority, are in exhaustion expressing PD-1 protein [68].
View Figure 23
In the chronic phase and in refractory shock, corticosteroid pulses have also been the solution, as blocking inflammation has reversed the unusual causes of vasoplegia, noting that patient COVID-19 has a lack of catecholamines, vasodilatation by adenosine and large production of nitric oxide [69,122,123].
Thus, the patient in inflammatory shock should maintain norepinephrine and vasopressin, dopamine (if possible all in low doses) and perform a corticoid pulse, with preference for Methylprednisolone.
COVID-19 is a mysterious disease, as it can simulate signs and symptoms of other known diseases, making its diagnosis difficult, especially in the chronic inflammatory phase of the disease called "Long COVID-19 Syndrome". The disease progresses with a viral infection for about 14 days, and the patient starts symptoms between the fifth and seventh days of infection; therefore, lung lesions and other infected organs have already occurred. After the seventh day of symptoms, few viruses remain, except those that may have predictors of severity (insulin resistance, age, and other inflammatory diseases). Between the sixth (D6) and the ninth (D9), the defervescence phase occurs - as also occurs in yellow fever, passing to the convalescence phase or to the toxemic/immune-mediated phase, responsible for the great aggravation that can lead to acute respiratory failure.
Proteomic and genomic studies have demonstrated the activation of innate immunity in patients who progress with severity, while adaptive immunity still plays a fundamental role against the virus in patients who evolve oligosymptomatic or asymptomatic.
CXCL8, CXCR1 and CXCR2 for neutrophil activation and accumulation, and inflammatory response genes (TLR4 and TLR6) associated with toll-like receptors and several significant inflammatory response genes (MMP8, MMP9, S100A12, S100A8), T cell activation, leukocyte-mediated cytotoxicity, natural killer (NK) cell-mediated immunity, and interferon (IFN)-gamma production were explicitly decreased in critically ill patients compared to groups that evolved without severity. Essential genes for T cell activation, such as CD28, LCK and ZAP70, and key transcription factors for the production of IFN-gamma (GATA3, EOMES and IL23A) are also low in critical patients. In addition, digital cytometry estimation revealed lower numbers of T and NK cells in critically ill patients. Protein polyubiquitination and autophagy increased gradually from asymptomatic to mild/severe and peaked in the critical group. An important coding factor gene for FOXO3 autophagy is also displayed in this expression pattern. Excessive activation of neutrophils can abnormally differentiate into pathological low-density neutrophils with an increased ability to release extracellular neutrophil traps (NETs). Excessive release of NETs causes endothelial damage, promotes thrombosis and contributes to mortality in COVID-19. Transcription analysis showed that neutrophils were significantly enriched in asymptomatic patients and slightly increased in critically ill patients. However, most proteins, including those involved in inflammatory pathways (CHI3L1, S100A8, S100A9, S100A11 and S100A12), neutrophil degranulation (ANXA3, FGL2, LRG1, PGLYRP1, DEFA1B and SLPI) and NETs (MPO and ELANE), were deficient in asymptomatic patients and increases progressively with disease severity. Notably, myeloid leukocyte activation and neutrophil degranulation pathways were enriched in these genes, further supporting the remarkable heterogeneity of neutrophils across different disease severity groups [21,28,50,60,61,150,157-160].
Try degradation products, deplete T cells, increase apoptosis of T helper (Th) cells and NK cells, and promote T cell exhaustion. L-arginine is essential for T cell proliferation and function, and the release of arginase (ARG1/2) from activated neutrophils inhibits T cell activation by inducing depletion of L-arginine and glutamine. ARG1 and ARG2 levels were up-regulated in critically ill patients. Consistently, L-arginine, N-acetylornithine and L-glutamine decreased in critically ill patients. Exhaustion markers, e.g. CTLA4, BTLA, HAVCR2, ICOS and PDCD1, were significantly up-regulated in critically ill patients. Multiple upstream IFN molecules, including TLR3, IRF1, IRF7, MAVS, DDX58, TBK1, JAK1 and STAT2, were down-regulated in symptomatic, especially critically ill patients.
Patients with severity predictors change the immune response to innate immunity with tryptophan metabolism facing the Kynureninne pathway - highly immunosuppressive - and based on interleukin 10 (IL-10) and regulatory T cells making the microenvironment tolerant, tumorigenic and immunoparalyzed. Monocytes/macrophages and DCs are concentrated in the production of IL-6, IL-10, TGF-1b, and neopterin, allowing the formation of the BH4 enzyme, whose function is to reduce the intensity of oxidative stress the formation of NO.
However, the BH4 pathway is compromised due to the action of Kynurenine products. Furthermore, the intestinal internalization of ACE-2 decreases the Try and Phe supply, negatively impacting the formation of catecholamines and serotonin [40,67].
The action of Patients with high serum 5-HT levels will have even more severe immunosuppression with neutrophilia and hyperthermia due to thermogenesis. Thin patients without insulin resistance or inflammatory comorbidities evolve with hypothermia and leukopenia resulting from the acute lack of serotonin. These facts are dependent on the inoculum to which the patient was exposed and gastrointestinal involvement by SARS-CoV-2. Furthermore, the permanence of a hypoxemic environment with adenosine production allows for a further shift of immunity towards the axis of tolerance and bradycardia [94,96,161].
Neurological symptoms result from acute lack of serotonin, excess of Kynurenine products and excess of Phe and adenosineph [32,68,73,118,155,162-166].
The oxidative stress axis is fed by multiple pathways: more inflammatory environment due to internalization of ACE-2, hypoxemic environment, impediment of the action of BH4, consumption of NAD/NADH+. Thus, the signature of inflammation in critically ill patients is represented by innate immunity, serotonin and oxidative stress (Figure 24).
 Figure 24: The figure represents immunosuppression pathways activated by SARS-CoV-2 and the impact of Kynurenine products inhibiting BH4, allowing more oxygen reactive formation due to the uncoupled form of NOS.
Figure 24: The figure represents immunosuppression pathways activated by SARS-CoV-2 and the impact of Kynurenine products inhibiting BH4, allowing more oxygen reactive formation due to the uncoupled form of NOS.
Hypoxia-associated accumulation of extracellular adenosine might be a critical immunoregulatory signal. A2 receptors for extracellular adenosine might act as primary sensors of excessive collateral tissue damage during an immune response and trigger the emergency downregulation of overactive immune cells.
Cathepsins activate innate myeloid immune cells since they contribute to toll-like receptor signalling and cytokine secretion. Furthermore, they control lysosomal biogenesis and autophagic flux, thus affecting innate immune cell survival and polarisation. External and internal stimuli, such as reactive oxygen species (ROS), lysosomotropic agents, certain chemotherapeutics, destabilise lysosomes and provoke lysosomal membrane permeabilisation. Leaked cathepsins (B, H, L, K, and S) trigger apoptosis by activating pro-apoptotic Bid and degrading anti-apoptotic Bcl-2 family proteins, promoting the release of mitochondria C cytochrome and engaging caspases.
Inflammatory cytokines increase the enzymatic activity of the indoleamine 2,3-dioxygenase (IDO) in activated monocytes, macrophages and brain microglia in inflammatory conditions. The essential amino acid tryptophan is therefore used to synthesise kynurenine, and this is at the expense of the synthesis of monoamine serotonin that depends directly on the availability of its precursor and limiting factor tryptophan. IDO activation might therefore impair serotonin neurotransmission. Kynurenine is then used to produce different neuroactive glutamatergic metabolites, including kynurenic acid, which is neuroprotective by acting on both glutamatergic NMDA and α7-nicotinic acetylcholine (α7-nAChR) receptors, and 3-hydroxykynurenine and quinolinic acid that are rather neurotoxic by oxidative stress or activating NMDA receptors. The activity of the kynurenine monooxygenase (KMO) that synthesises 3-hydroxykynurenine is increased in activated microglia by inflammatory cytokines, increased production of kynurenine is ultimately associated with skewing of the kynurenine metabolic balance toward increased neurotoxicity. By impairing serotonin neurotransmission and promoting neuronal damage, cytokine-induced kynurenine pathway activation may contribute to inflammation-driven depressive symptoms. Increased xanthurenic acid, a metabolite of 3-HK, has been shown to directly lower BH4 biosynthesis by inhibiting sepiapterin reductase. Similarly, it has been suggested that the concurrent upregulation of kynurenines and BH2 production may lead to a combined up-regulated activity of NOS (by kynurenines) and decreased availability of BH4, the NOS cofactor.
Inflammatory cytokines, including IL-1β, interferon-γ and TNF-α, can induce GCH1 expression and activity, increasing BH4 synthesis. At the post-translational level, GCH1 is inhibited by BH4 and stimulated by phenylalanine through its complex formation with the cyclohydrolase feedback regulatory protein (GFRP). BH4 is also oxidised to BH2, which can be converted back to BH4 by the dihydrofolate reductase (DHFR).
In the BH4 salvage pathway, eNOS cofactor BH4 is readily oxidised to BH2 in circulation. In cases of decreased BH4 bioavailability, eNOS uncoupling occurs, and eNOS produces ROS instead of NO. BH2 may be reduced back to BH4 via folate dependent DHFR via the BH4 salvage pathway. This pathway maintains an adequate BH4: BH2 ratio to support NO production. It is us; endothelial nitric oxide synthetase, BH4; tetrahydrobiopterin, BH2; 7, 8-dihydrobiopterin, DHFR; dihydrofolate reductase.
Nitric oxide synthase (NOS) activity. NOSs are homodimers, and each monomer contains a reductase domain and an oxygenase domain connected by a calmodulin-binding peptide linker (CaM) (for eNOS and nNOS). In the normal NOS coupled state, electron (e−) flow through the nicotinamide adenine dinucleotide phosphate (NADPH) and flavin domains, adenine dinucleotide (FAD), and flavin mononucleotide (FMN), from the reductase domain of one monomer to the iron of the heme group (Fe) localised in oxidase domain of the other monomer. (6R)-5,6,7,8-tetrahydrobiopterin (BH4) binding to the heme active site at the interface between the two monomers increased L-arg (L-arginine) substrate interaction and dimer stabilisation, producing nitric oxide (NO) and L-cit (L-citrulline). In the uncoupled state caused by the absence of BH4, the electron transfer and the reduction of oxygen (O2) are uncoupled from L-arginine oxidation resulting in the generation of superoxide anion (O2•−).
View Figure 24
COVID-19 is a viral disease that has caused many disorders in people's lives. It is an absolute catastrophe in both the acute and chronic phases. The acute phase still has the same treatment protocols since the beginning of the pandemic, and patients continue to evolve to death. Although we still do not have the results of large trials, studies from research centres and quality hospitals show the benefit of using some drugs, such as selective serotonin reuptake inhibitors. Furthermore, recognizing thermogenesis allows its treatment with an insulin pump. At a critical moment such as this pandemic, ethical issues must be related to the intention to do something different; however, it is based on profound and reliable studies. It is not unethical to use a drug as long as it is well studied and supported by the literature when people are dying in the same way.
The chronic phase, "The Long COVID-19", has been an unprecedented catastrophe. Patients, mainly the elderly, are admitted to hospitals with acute delirium or with an acute change of consciousness without image changes in computed tomography of the skull. However, when researched, it is common to find pulmonary images of previous COVID-19 sequelae, increased LDH and a generalized inflammatory profile with urine showing proteinuria and hematuria, sometimes with ketone bodies. This condition is often associated with weight loss. Usually, these patients have been diagnosed with some neoplastic disease-causing wasting syndrome (this can occur since the patient is immunosuppressed by COVID-19). However, the condition can often be reversed using corticosteroids and vitamin B complex replacement, L-Dopa replacement, and the use of serotonin reuptake inhibitor.
I have been following patients undergoing "The long COVID-19", and the complaints are the same: pain in the calves, hair loss, exacerbation of pulmonary or upper airway conditions, or abdominal conditions ranging from tenesmus and diarrhoea. These conditions have had an excellent response to the use of corticosteroids, ondansetron and tryptophan and B complex replacement. I always tell patients that everything is still theoretical and that I also have many doubts, so we always build the therapeutic plan together. Nevertheless, I reinforce that the patient is always well informed about everything, including my doubts about something new like SARS-CoV-2. So far, I have not had cases that worsened due to the use of these drugs; on the contrary, there was an essential improvement in symptoms, improving patients' quality of life.
We have to be careful because in a pandemic, mathematically, those who are not infected will still become infected, and, in the case of SARS-CoV-2, those who are already infected can become infected again. Thus, COVID-19 should be included in the diagnostic hypotheses whenever conditions might suggest an underlying inflammatory cause or something new that did not exist in the patient before. The start could have been COVID-19.
We cannot see SARS-CoV-2 as simple. This new adversary needs to be studied and well understood to act in the best possible way and the consequences left by him in people. We have to recognize our ignorance and study because our knowledge is limited and to think that we already have all the answers to the unknown is to be anti-science. It is stopping the search for knowledge since what drives us are doubts and questions.
In the beginning, the article proposed the pathophysiology of this probable COVID-19 based on news publications showing the presence of tryptophan metabolites via the immunosuppressive pathway of Kynurenine in patients who died from yellow fever. The clinical changes seen over almost two years of follow-up of COVID-19 patients, aided by laboratory tests, allow the comparison between the two diseases, reinforced by neuropsychological and immunological clinical symptoms and by laboratory tests that, in greater magnitude, show neutrophilia in patients who progress to death in both diseases.
These latest published studies helped to understand a possible explanation for the evolution of the disease in obese, diabetic and elderly patients, especially.
It turns out that, unlike Yellow Fever, COVID-19 perpetuates a much more exuberant and prevalent inflammatory status, activated by multiple pathways and feedback.
Chronic COVID-19 is a Thrombo-metabolic Syndrome, tolerant and immunosuppressive, whose intensity varies from person to person, but it is more intense in critically ill patients or who were once in patients in ICUs. Chronic inflammation can appear at any time - near the acute phase or farther away. It is essential to understand if the patient is inflamed in its entirety, presenting systemic changes. "The Long COVID-19" mimics bacterial sepsis but responds poorly to the use of antimicrobials. It is necessary to stop the inflammation using corticosteroids, but antimicrobials are necessary due to the intense bacterial translocation through the intestine and the skin.
The pulmonary image is usually sequelae with pleural effusion and posterior atelectasis in the lung bases, laminar atelectasis, consolidations forming something like nodules or images confused with fungal or mycobacterial infection. This type of formation is seen in rheumatologic and inflammatory diseases and is called organizing pneumonia.
Diagnosing "The Long COVID-19" is difficult, but few tests can help with the diagnosis.
|
Recommendations:
Image: Chest computed tomography showing the presence of organizing pneumonia or eosinophilic lungs or minor sequelae such as laminar atelectasis and small effusions in the posterior and lung bases.
Laboratory: 1. Complete blood count: observe monocytosis, lymphopenia and eosinophilia that may be mild due to migration of these cells to tissues. 2. LDH: Elevated, showing anaerobic respiration. 3. Urea: Usually elevated by the mobilization of amino acids to perform gluconeogenesis. 4. Creatinine: Normal to elevated. 5. General urinalysis: The presence of variable proteinuria accompanying disease progression or decreased proteinuria; when the inflammatory disease improves, hematuria, leukocyturia, sometimes ketonuria, with the presence of bacteria or fungal filaments, sometimes with the presence of casts. 6. Usually elevated C-reactive protein. 7. Glucose: Slightly elevated to very high. 8. Triglycerides and Cholesterol: Slightly elevated to very elevated. 9. Platelets: Follows a sinusoidal profile of fall and rise.
Clinical Phenomena 1. Delirium. 2. General changes in consciousness regardless of age, but more prevalent in the elderly. 3. Depressive, anxious or schizophrenic symptoms. 4. Age-independent loss of cardiac function. 5. Increased cardiac area. 6. Anhedonia. 7. Infection (or inflammation) of the recurrent urinary tract. 8. Weakness and pain. 9. Visual loss. 10. Abdominal symptoms: ulcerative colitis-like lesions, tenesmus or diarrhoea. 11. Exacerbation of pulmonary or upper airway respiratory conditions. 12. Skin lesions, skin rash. |
To COVID-19 patients who have chronic inflammation of the disease and who remain discredited and neglected by the government. Thank you for the learning I have with you daily.
I, Luiz Zanella, wrote this article and elaborated the figures based on the Biorender platform. I have been performing an active participation in the management of COVID-19 patients.
The author has not competing interests.
Pelletier, et al. [1] show that in Yellow Fever disease that aromatic amino acids tyrosine, tryptophan and phenylalanine were enriched in patients with good follow up. The fact that COVID-19 has such an intense cytokine storm can be related to a deficit of Tryptophan and Phenylalanine as already published in previous works published by me, explaining this relationship of amino acids, ACE-2 and immunological response in COVID-19. From that theory and the work that makes it most justified, consider the key points below.
1. COVID-19 is a SARS-CoV-2 infection that triggers an automatic inflammatory feeding loop by tryptophan (Try) deficiency due to the internalization of ACE-2, causing an imbalance in the inflammatory pathway by the immune response against SARS-CoV-2 and the anti-inflammatory pathway mediated by Kynurenine (KYN).
2. Maintaining hypoxemia creates a tolerant and tumorigenic microenvironment.
3. Niacin is essential for aerobic respiration to occur. In its absence, the tryptophan pathway tends to alter the balance for the formation of B3. However, in the tryptophan pathway, inflammation, via IDO-1, tends to metabolize kynurenine by-products - toxic and immunosuppressive.
4. Several metabolic pathways suffer from SARS-CoV-2 interference causing neuropsychiatric symptoms (tremors caused by anxiety, akathisia, mental confusion, acute depression. For example:
a) Serotonin depletion
b) Toxic products of kynurenine metabolism
c) Eosinophils resulting from IL-6 stimulation in bone marrow
d) Post-viral infection that triggers autoimmune disease
One of the products of hypoxia is adenosine, whose effect is anti-inflammatory, and its overproduction due to severe hypoxemia may explain bradycardia as the main arrhythmia of COVID-19 and the myocardial damage caused by a cytokine storm.
5. Thrombosis in COVID-19 may be the primary cause of high serotonin levels (5-HT) in obese or glucose-resistant patients.
6. Thrombogenesis takes several ways, but it needs to act on the cause and not just on the phenomenon to overcome thrombogenesis.
Inflammation and hypoxemia must be blocked so that anticoagulant or antiplatelet therapies are not refractory.
7. These pathways (NAD/NADH+, expression of furins and cathepsins) show that we should not keep the patient hypoxemic, and sometimes IOT is mandatory.
8. The intense production of adrenaline stimulated by a highly stressful environment triggers an exacerbated thermogenesis after the consumption of serotonin, which starts to express UCP-1.
9. Thermogenesis produces tissue damage and chronic inflammation in patients with COVID-19.
10. Corticosteroids are still a good weapon against a disease that causes inflammation through multiple pathways. The strength of corticosteroids in COVID-19 lies in the fact that they are non-specific. Thermogenesis blockade is essential in the clinical management of critically ill patients COVID-19.
11. Neopterin, BH4, and Kynurenine are critical molecules for understanding the immunopathology in COVID-19.
12. Kynurenine, an anti-inflammatory pathway, has a paradoxical role in COVID-19, increasing oxidative stress.