The promising area of lipidomics has received a major interest as exploring panels of lipid biomarkers and their metabolic pathways might contribute to the early diagnosis of Alzheimer's disease allowing novel therapeutic targets. Dysfunctions in lipid metabolism have been associated with metabolic disorders as well as with aging and neurodegenerative diseases. Neurolipidomics is beginning to provide new insights into the pathophysiology of AD through identification of plasma and cerebral lipid mediators and metabolites (lipidome). Altered lipid profile and dysregulated metabolism of DHA, plasmalogen, phosphoinositol (PI), phosphoethanolamine (PE) has been investigated mainly through their incorporation into neuronal membranes and may all contribute to AD progression. This review summarizes the current knowledge on altered lipidome in AD and the impact of dietary lipids (cholesterol, phospholipids, SF, LA, AA, EPA, DHA) on the brain, CSF and plasma lipidome in MCI and AD subjects compared to age-matched controls.
Lipidomic analyses suggest that altered lipid pathways in brain and peripheral tissues may affect the progression of the disease [1]. Altered lipid metabolites and dysregulations in cerebral lipid homeostasis have been associated with onset and progression Of AD. Lipid software programs and databases (Lipidmaps; LipidBank; LipidXplorer) have been developed for quantification of lipid classes, identification of physicochemical characteristics, data analysis and biochemical pathway restoration. Phospholipids (PL) that account for 20-25% of the adult brain's dry weight are mixed acid esters of glycerol with two fatty acids and one phosphoric acid moiety [2]. Derivatives of phospholipids are cellularly synthesized de novo from a-glycerol phosphate, fatty acids and other molecules esterified to phosphatidic acid. Phospholipids can also be provided through dietary sources (egg yolks, soya, meat, fish, milk and dairy products). Glycerophospholipids (GPL) and sphingolipids are the two major classes of phospholipids and both have been associated with neurocognitive health. The backbone of GPL is glycerol attached to three hydroxyl groups named as sn-1, sn-2 and sn-3. The attached phosphate group at sn-3 position may be either choline, etholamine or serine and give rise to PC, PE and PS respectively. GPL might contain n-3 fatty acids in the form of DHA if derived from DHA rich marine foods (krill, squid) or from eggs fortified with omega-3 FA foods [3]. Palmitoyl CoA and serine condense to form dehydrosphingaganine which in turns converted to sphingosine. It then reacts with a long chain acyl-CoA to form ceramide. The terminal hydroxyl group is substituted by phosphorylcholine to form sphingomyelin [4]. Lipid rafts are mainly composed of sphingomyelin, glycosphingolipids, and cholesterol. Sphingolipids, glycerophospholipids and cholesterol play a crucial role in many cellular functions including cell membrane formation, energy storage and cellular signaling [5].
Through neurolipidomic datasets, altered phospholipid metabolism in neuronal membranes has been detected. Plasmalogens are a subclass of glycerophospholipids that possess a vinyl ether fatty alcohol substituent at sn-1 of the glycerol backbone. Contents of ethanolamine plasmalogen, choline plasmalogen [6], phosphatidylinositol (PI) and phosphatidylethanolamine (PE) are all reduced in the brain tissue samples of AD subjects compared to controls, designating a high phospholipid turnover in AD brain. On the other hand, significantly higher levels of serine glycerophospholipids have been observed in AD [7], supporting the hypothesis that membrane phospholipid alterations might be involved in signal transduction abnormalities in AD. Accumulated levels of PC(O-16:0/2:0) and PE(P-16:0/0:0)] was associated with tau pathology, signaling neuronal dysfunction and loss. On the other hand, reduced levels of PI(16:0/20:4), PI(16:0/22:6), and PI(18:0/22:6) contributed to Aβ42 biosynthesis. These metabolic dysruptions are critical for the conversion of the pre-symptomatic to the symptomatic stage of AD [8]. As indicated by several studies, a depletion of PUFAs and increased levels non-esterified monounsaturated fatty acid (MUFA) in AD brains have been observed [1]. Shotgun lipidomics techniques revealed reduced levels of Sphingomyelins (SM) and increased ceramide concentrations in AD brains due to SMase activation and the consequent SM hydrolysis into ceramide production. Reduced sphingomyelin SM content in the middle frontal gyrus of AD patients when compared to controls, [9] whereas other post mortem analysis techniques demonstrated opposite results with, SM be overexpressed in n middle frontal gyrus gray matter.
Glycolipids are conjugates of carbohydrates (glucose, galctose) with N-acetylglucosamine and N-acetylneuraminic acid with sphingosine. They are common membrane components of the central nervous system and peripheral nerve tissues. Glycolipids are mainly ceramides derivatives and play a regulatory role in membrane trafficking, signal transduction, cellular adhension and recognision. Subclasses of glycolipids include neutral glycoshingolipids (cerebrosides), Sulfoglycosphingolipids (also called sulfatides) and gangliosides. The latter is found abundantly in diet (eggs, meat, milk) and it is crucial for brain development. The content of ganglioside is significantly depleted during aging and in brain areas that most affected by AD. Concentrations of ganliosides of the ganglio-series (GM1) become diminished especially in the frontal cortex, temporal cortex and white matter whereas simple gangliosides (GM3) are accumulated in the aged frontal and parietal cortex [10]. Interactions between lipid rafts and Aβ may promote AD pathogenesis whereas another study suggests a possible conformation of Aβ directly after its binding to the ganglioside GM1 [11].
Sulfatides or 3-O-sulfogalactosylceramide is a major component of myelin sheath in the peripheral and central nervous system. Alterations of brain sulfatide levels and abnormal sulfatide metabolism might have an impact on the pathophysiology of both AD and Parkinson's. Reduced sulfatide levels in grey and white matter were found in AD subjects, using electrospray ionization mass spectrometry [12]. This reduction might be associated with the expression of ApoE ε4 allele, since metabolism of ApoE modulates the lipid content in the brain [13].
Considering the essential fatty acids, docosahexaenoic acid (22:6n-3, DHA) and arachidonic acid (20:4n-6, AA) account for about 8% and 6% of the dry weight of human brain, respectively [14]. Polyunsaturated fats serve as modulators of neuronal membrane fluidity and as precursors of lipid mediators that regulate inflammatory response. Both linolenic (ω-3FA) and linoleic acids (ω-6FA, sunflower oil) are essential and should be provided through diet since humans and animals cannot synthesize them. Linoleic acid is metabolized to γ-linolenic acid (GLA,18:3n-6), which is then elongated to dihomo-γ-linolenic acid (DGLA) and eventually is metabolized to arachidonic acid. N-6 fatty acids are precursors of eicosanoids such as prostaglandins, thromboxanes, leukotrienes, lipoxins, resolvins, eoxins [15]. GLA, even at low doses showed inhibitory effects on the formation of advanced glycation end products (AGEs) that lead to memory impairement. Administration of GLA (1 and 5 mg/kg) significantly attenuated the effect of d-fructose on the HbA1c level in an animal study. Interestingly, an in silico study confirmed interactions between GLA and active site residues of the RAGE receptor. The GLA-RAGE interactions exhibited hydrogen bonds with Arg77, and Gln67 residues and hydrophobic interaction with Try 72 of the binding site of RAGE [16]. Moreover, a blend of evening primrose oil (linoleic,GLA)/fish oil (EPA, DHA) with ratio of omega-6 to omega-3 fatty acids of 2.06:1 reduced progression of aliminium-induced AD in rats. The GLAs in EPO give rise to anti-inflammatory eicodanoids. Dihomo-γ-linolenic acid (DGLA), an important constituent of neuronal membranes is stored as phospholipids serving as a precursor of prostaglandins designated E1 through cyclooxygenase [17]. Arachidonic acid (C20:4,AA) is also stored within the phospholipid fraction of cell membranes, where it acts as a precursor to the family of prostaglandins designated E2. AA contrary to GLA contributes to AD progression, since upregulation of AA cascade has been observed in AD patients.This hypothesis was confirmed through measuring mRNA levels of AA with neuroinflammatory markers such as IL-1β;TNF-α;PGE2 in postmortem frontal cortex from AD patients and matched controls. Thus,the enhanced consumption of n-6 PUFA leads to an excessive production of the proinflammatory cytokines derived from AA through COX and LOX enzymatic activity which lead to brain damage.
A multicentre cohort study from the German Study on Ageing, Cognition and Dementia in Primary Care Patients (AgeCoDe) with a 7-year follow up period, was conducted in order to investigate associations between fatty acid profile in serum phospholipids and incidence of all-cause dementia and AD. After controlling for various confounders including age, gender, education, APOE ε4 status, BMI, smoking, physical activity, vitamin E, total cholesterol, triglycerides, lipid lowering medication, the study revealed an inverse association between serum phospholipid EPA and the incidence of dementia AD [18]. In agreement with these results, administration of EPA-enriched phosphatidylethanolamine (EPA-PE) and especially eicosapentaenoic acid (EPA)-enriched ethanolamine plasmalogens (EPA-pPE) in a rat model of AD, attenuated Aβ-induced neurotoxicity, reduced GSK-3β, tau phosphorylation and oxidative stress [19]. Incubation of neuronal cells with docosahexaenoic acid (DHA, 22:6n-3), enhanced PS (phosphatidylserine) incorporation in neuronal membranes and demonstrated a considerably reduction of DNA fragmentation and a downregulation of caspase-3 activity, both indicators of reduced apoptic cell death [20]. The Etude du Vieillissement Artériel (EVA) cohort study with a 4-year follow-up design, revealed an inverse association between cognitive decline and DHA concentrations in erythrocyte membranes, as opposed to stearic acid levels [21]. Similarly, other prospective and observational studies confirmed the protective role of regular fish consumption (weekly) and dietary intake of DHA against AD [22]. Dietary n-3:n-6 PUFA ratio alters the fatty acid composition of membrane phospholipids and neural membrane PUFA composition has an impact on membranous fluidity, gene transcription, neural apoptosis and death.The balance between the n-6:n-3 PUFA ratio may therefore play a crucial role in the onset of AD. Such alterations in the FA composition of erythrocyte takes place in early pro-symptomatic AD. Individuals with neocortical beta-amyloid load (NAL) demonstrated elevated AA and low docosapentaenoic acid (DPA) concentrations in plasma [23]. Albeit liver's capacity to synthesize DHA from a-La in order to supply DHA brain levels has been reported in considerable amount of research [6,24,25], it should be mentioned that liver peroxisomal function of AD subjects becomes inadequate to convert tetracosa hexanoic acid into DHA with subsequent reduced brain DHA levels [1]. Other studies highligheted that the reduced liver fatty acid desaturase capacity in combination with age-dependent defects in endogenous antioxidant defense systems may give rise to lipid peroxidation [26]. Under these conditions, peroxidation vulnerability of long chain PUFAs due to the presence of double bonds, especially DHA, should be considered before they are given as neurotherapeutic regimen. Moreover, neuroprotectin D1 (NPD1;10R,17S-dihydroxy-docosa-4Z,7Z,11E,15E,19Z hexaenoic acid, a DHA-derived lipid mediator, downregulates oxidative stress-induced caspase-3 activity and inhibits apoptosis through attenuation of proapoptic Bcl-2 family members Bax and Bak expression [27]. This DHA-derived docosanoid has been shown to be neuroprotective since contributes to the regulation of circuit integrity, neuroinflammatory signaling and synaptic homeostasis when disruptions and neurodegenerative mechanisms occur [28]. NPD1 inhibits amyloidogenic processing of APP and attenuates pro-inflammatory gene expressions including TNF-a and cyclooxygenase-2 [29]. DHA and DHA-derived lipid mediators exert antioxidant, anti-inflammatory and antiapoptic actions. Both DHA and NPD1 are underexpressed at the hippocampal cornu ammonis region 1(CA1) from AD patients [30,31].
Cholesterol is the most widely distributed sterol in animal and human tissues. This simple lipid is a major constituent of the human brain accounts for 20% of the body's total cholesterol and and as free cholesterol is present in central and peripheral nervous system. Cholesterol is a crucial molecule for brain development, synaptogenesis and synaptic plasticity, dentrite formation and proper neurotransmission. However, increased levels of circulating cholesterol may contribute to the incidence of sporadic AD. From epidemiological perspective, hypercholesterolemia as well as the inheritance of the epsilon 4 allele of the ApoE gene represent major risk factors for AD. Cholesterol has a direct impact on activity of β- and γ-secretases and could modulate bilayer thickness affecting the cleavage activity and specifity of γ-secretases for Aβ40 species rather for pathogenic Aβ42/43 species [32].
High cholesterol diets increase cholesterol circulation in brain parenchyma and cause the aggregation of free cholesterol in neurons of mice, rats and rabbits [33,34]. This cholesterol build-up changes both structure and function of endolysosomes resulting in significant depositions of Aβ and phosphorylated tau in olfactory bulb neurons [35]. Cholesterol fed rabbits, 1% or 2% for 6 or 12 weeks respectively came up with increased levels of AB in hippocampus and cortex, as demonstrated with immunohistochemisty and ELISA assays. Although a systematic review with Meta-Analysis failed to show any significant association between total serum cholesterol-at midlife-and cognitive impairment, other researchers support the theory that elevated plasma cholesterol may contribute to the pathogenesis of AD, through different mechanisms. As indicated in previous studies, BBB plays a crucial role in homeostasis of the brain microenvironment and dysfunction of BBB precedes neurological diseases such as stroke and AD. Following a high fat diet for 12 weeks and a high cholesterol diet, the BBB leakage of mice showed respectively a 30-fold deterioration and a 7-fold greater dysfunction compared to control ones [36].
Οxysterols, mainly known as 24S- and 27-OH cholesterol, are oxidized metabolites of cholesterol that cross the blood brain barrier [37]. 24S-OH can be transferred from the brain into the circulation whereas 27-hydroxycholesterol can enter the brain at the level of 4-5 mg each day [38]. 24S-hydroxycholesterol is a steroid derivative which crosses the blood brain barrier and is converted enzymatically via cholesterol 24S-hydroxylase from CNS cholesterol. Large, longitudinal population studies [39] have underlined an increased ratio of 27-OH cholesterol to cholesterol as a predictor of worsening cognitive impairement. Furthermore, using combined gas-chromatography/mass spectrometry methods, researchers [40] determined increased plasma and CSF levels of 24S-hydroxycholesterol in AD and MCI patients. There is speculation that changes in cholesterol metabolism may be involved in the pathogenesis of AD, and as a result the metabolite of 24S-OH-Chol could be used as a potential biomarker for the progression of the early stages of the disease. High levels of free cholesterol measured in middle frontal gyrus of AD patients [9] and a number of oxysterols derived from cholesterol auto-oxidation such as 7-ketocholesterol,7α-hydroxycholesterol, 4β-hydroxycholesterol, 5α,6α-epoxycholesterol, and 5β,6β-epoxycholesterol have been identified in post-mortem human AD brains, during the disease progression [41].
High dietary intake of saturated fats enhances the brain fatty acid uptake from plasma as indicated via positron emission tomography (PET) by Karmi, et al. [42]. Saturated fatty acids lead to overexpression of pro-inflammatory cytokines (IL-1, IL-6 and TNF-α) mediating neuroinflammation in astrocytes and microglia and causing neuronal death [43,44].
Ceramides are lipid mediators and common components of all glycolipids which are common membrane components of the central nervous system as well as the precursors of sphingomyelin and gangliosides. They act as bioactive molecules in several cellular pathways including apoptosis and intracellular signalling [45]. Ceramides are synthesized de novo, in the endoplasmic reticulum (ER) by the reaction of a long-chain acyl CoA with sphingosine. Ceramide is then transported from the ER to the Golgi apparatus for the formation of sphingomyelin [46]. Acid sphingomyelinase (ASM), a lysosomal glycoprotein cleaves sphingomyelin into ceramides and acid ceramidase further metabolizes ceramides to sphingosine and sphingosine-1-phosphate [47]. Diabetic models, upon a high fat diet have also demonstrated increased levels of circulating ceramides. Oversupply of dietary fat in the form of saturated fat has been shown to affect ceramide metabolism leading to lipotoxicity, membrane reorganization and cellular dysfunction. Most experimental animal studies indicate that a high fat diet in combination with an excessive palmitate intake through ingestion of palm oil lard, butter, cheese, lamp, pork loin, salami, goose, chicken with skin appear to increase neurotoxic ceramide concentration in different tissues [48]. Moreover, astrocytes when treated with palmitate contributes to the secretion of IL-1β , BACE 1 and Aβ42 in neuronal region [49].
The impact of high palmitate-diet regarding activation of NF-κB on mouse hippocampus was identified [50]. Treatmenent with palmitate increased the expression of bace1 and amyloidogenic pathway of ΑβPP via NF-κB transcriptional activity. In another animal study, exposure of hippocampal and microglia cultures to palmitate, contrary to oleate [51] induced microglia activation as well as cognitive impairment via TNF-a activation and respectively caused hippocampal astrogliosis [52] (Figure 1). It has also been suggested that elevated levels of blood ceramides in amnestic MCI subjects had a predictive value for cognitive deterioration and hippocampal volume loss [53].
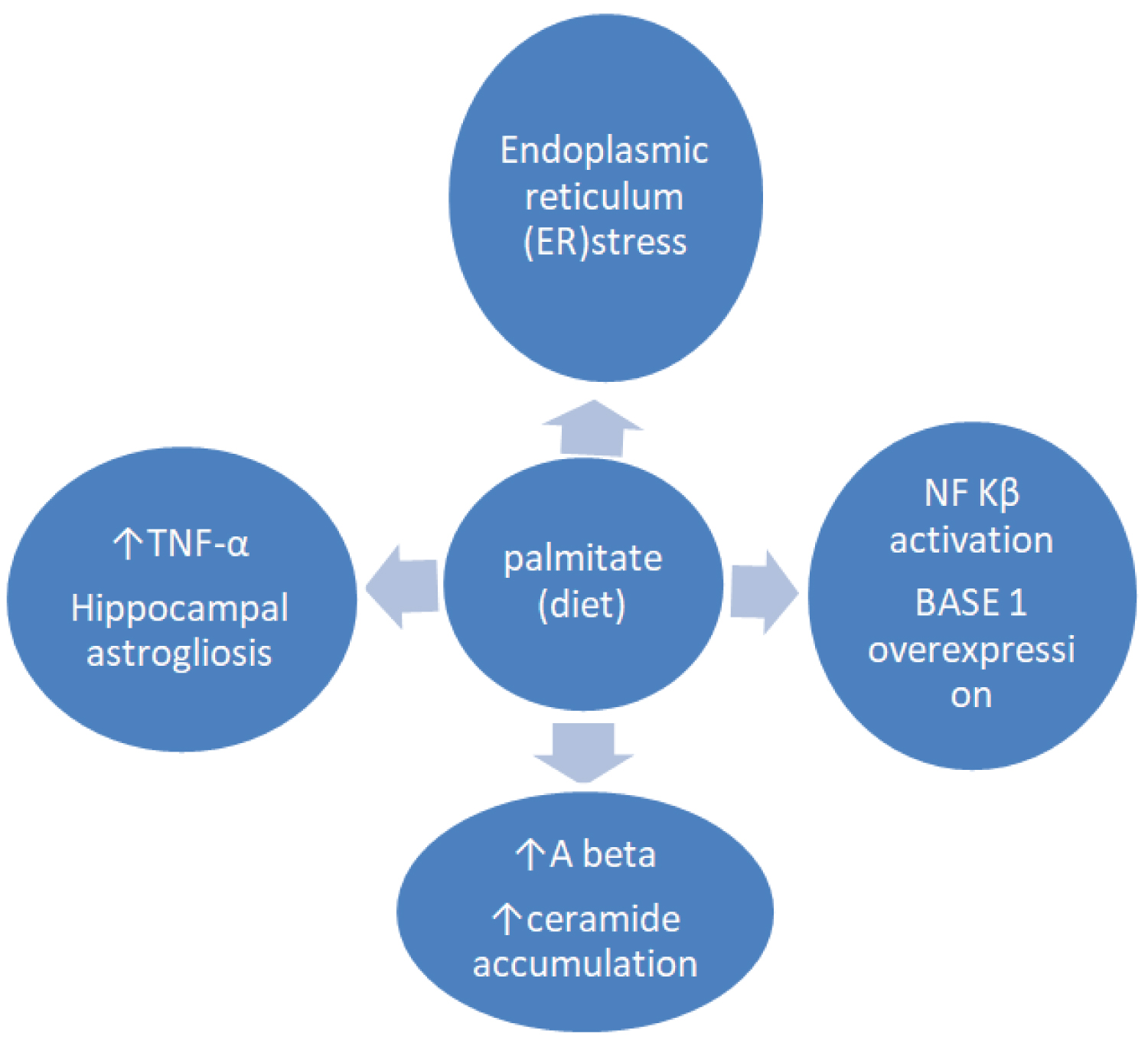 Figure 1: Exposure to high saturated fat diet (palmitate) promotes endoplasmic reticulum (ER) stress, triggers proinflammatory cytokine production, hippocampal astrogliosis, BASE 1 overexpression via NF Kβ activation and Aβ accumulation eventually leading to the progressive Alzheimer's disease.
View Figure 1
Figure 1: Exposure to high saturated fat diet (palmitate) promotes endoplasmic reticulum (ER) stress, triggers proinflammatory cytokine production, hippocampal astrogliosis, BASE 1 overexpression via NF Kβ activation and Aβ accumulation eventually leading to the progressive Alzheimer's disease.
View Figure 1
Saturated fat-enriched diets, contrary to PUFAs result in fatty liver synthesis and enhanced circulating ceramides [54], which in turns contribute to insulin resistance,that triggers AD pathogenesis [55]. Oversupply of SFA (palmitate) leads to accumulation of ceramides that impair insulin signaling pathways and suppress the activity of PKB/Akt [56]. Other studies based on enzymatic assays also confirmed an altered brain sphingolipid metabolism and brain overexpression of ceramide, sphingosine, glycosylceramide synthase, and membrane-associated acidic ceramidase and sphingomyelinase activities in AD or mild cognitive impairment compared to controls [57,58]. On the other hand, when mouse MIN6 insulinoma cells treated with olaeate, insulin sensitivity was enhanced through inhibition of ceramide species biosynthesis [59]. Accumulation of ceramides is capable of inducing apoptosis in cortical and hippocampal neuron cultures through p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) phosphorylation [60].
The F-ring isoprostanes (IsoPs) are synthesized by the peroxidation of the n-3 PUFA, EPA and DHA. Neuroprostanes which are oxidised metabolites of DHA are found abundantly in neuronal membranes. Lipid peroxidation and oxidative injury occur early in amnestic mild cognitive impairement. Accumulation of f4-neuroprostanes were found in parietal and occipital lobes in MCI and in late AD compared to normal control subjects [61]. Therefore, quantification of F4-neuroprostanes (F4-NeuroPs) can be used as a sensitive indicator/biomarker of oxidative damage in neuronal membranes in AD compared to F2-isoprostanes (F2-IsoPs). High susceptibility of DHA to oxidation and autooxidation process have been demonstrated in both in vitro and in vivo studies [62]. However according to the authors, dertermination of F2-IsoPs levels in CSF could not be utilized as an early marker of dementia. Resolvin and maresin also termed neuroprotectins are oxylipins or oxygenated derivatives of eicosapentaenoic acid (EPA), docosapentaenoic acid (DPA) and especially of docosahexaenoic acid (DHA) [63]. These molecules together with neuroprotectin D1(NPD1) promote the resolution of inflammation and reduce the risk of neuroinflammation-induced AD. They are also known as specialized pro-resoving lipid mediators (SPMs) and they are diminished in the CSF of AD subjects [64] (Table 1).
Table 1: Overview of altered lipidome and expression of lipid classes and lipid mediators in Alzheimer's disease. View Table 1
Altered plasma and brain phospholipidome as well as altered FA composition in lipid rafts within the cell membranes have been implicated in the progression of AD. The majority of neurolipidomics studies demonstrated dysregulated metabolism of DHA, plasmalogen, phosphoinositol, phosphoethanolamine (PE). Elevated expression of arachidonic acid (AA) in grey matter, ganglioside GM3, ceramides, 24S-hydroxycholesterol with neurotoxic effects have been observed in brain tissue, CSF or plasma. On the other hand, decreased levels of EPA, DHA, and DHA-derived neuroprotectin D1, maresin1, resolvin D1,lipoxin A4, sphingomyelins, sphingosine-1 phosphate, ethanolamine plasmalogens and sulfatide have been associated with AD progression. Saturated fat enriched diets incorporated into neuronal membrane phospholipids and eliminated levels of PUFA may lead to ER stress, ceramide generation, Aβ accumulation and hippocampal astrogliosis. Finally, palmitate through ingestion of butter, cheese, milk, beef, palm oil, might also trigger pro-inflammatory cytokines (TNF-a) and BASE1 activation. Understanding the role of dietary lipids on brain's phospholipidome in different stages of AD is a crucial step for the development of non-pharmacological therapeutic strategies. Considerable efforts should focus on the metabolic pathways of lipid mediators in different regions of the brain that still remain unexplored. The combination of neurolipidomics with genomics/proteomics in AD is a critical future area of research for early identification of biomarkers and personalized therapeutic strategies.