Stimulation of oral Type II taste cells with T1R2/R3 receptors elicits sweet taste and invites consumption. Intestinal Type II taste cells with T1R2/R3 receptors facilitate glucose absorption. Type II taste receptor cells contain a calcium channel, CALHM1, which if deleted results in loss of ability to sense and perceive the sweet taste quality. Comparison between mice with (+/+; WT) and without (-/-; KO) CALHM1 provides the means to examine T1R2/R3 receptor effects on intake and intestinal absorption via measurements of body weight (BW), blood glucose (BG) and plasma insulin. In this study we confirm our previous findings that WT mice are heavier, eat more, and have higher mortality than KO mice [1]. We report that higher BG and insulin levels accompany higher BW in both WT and KO mice, although, KO mice with the same BW as their WT counterpart have lower BG and insulin levels. Glucose gavage increased and prolonged BG and plasma insulin levels more consistently in WT than KO mice. Fructose exerted little effects on BG or insulin. Gavage with the high potency, non-saccharide sweetener SC 45647 had no effect on BG or insulin of KO mice, but caused some increase of both BG and insulin levels in the WT mice. The effect on insulin and BG by water gavage was negligible compared to that of glucose. These results suggest that inhibition of T1R2/R3 receptors lowers oral intake and intestinal uptake, which then results in lower BG and insulin levels. These findings can be applied to weight control in humans.
Mouse, Obesity, Diabetes, Insulin, Blood glucose, Sweeteners, CALHM1, T1R2/R3
Sweet taste is an innate taste quality linked to consumption [2-4]. It encourages over-consumption beyond basic needs and obesity has become one of the most prevalent problems in the Western world. Obesity permeates all facets of life with negative consequences from daily discomfort to prolonged health effects, including increased risk of diabetes cf [5-8].
Sweet taste is the result of stimulation of T1R2/R3 receptors on type II taste cells (TRC) in taste buds of the oral cavity [9]. These TRCs contain several other structures necessary for their function. One is a TRPM5 channel [10-13]. Another is a calcium channel, CALHM1 [14], which releases ATP transmitter to purinergic receptors on taste fibers [15]. Deletion of CALHM1 abolishes the nerve response to sweet stimuli in the chorda tympani [16] and glossopharyngeal nerves fibers [1]. Hierarchical cluster analyses, which takes into consideration all responses of a large array of tastants have identified the fibers that respond to sweeteners as a separate group called dedicated or specific S fibers. S fibers are present in humans [17-20], have been identified in nonhuman-primates [21-28] as well as in mice and other rodents [29-35]. In all these species S fiber impulses stimulate intake.
Our first study described the taste effects of deletion of CALHM1. It showed that mice with deleted CALHM1 (KO) were not attracted to sweet and showed no response to sweeteners in the chorda tympani nerve [16]. In a second study we reported the same effects on the glossopharyngeal nerve response as in the chorda tympani, combined with the finding that the KO mice were lighter, leaner and more active, with lower mortality than their normal tasting littermates (WT) mice [1]. This led us to this study of the possible role of intestinal sweet T1R2/R3 receptors on intake and body weight.
The present understanding of intestinal effects of sweeteners in general and glucose in particular is that intestinal glucose absorption occurs through enterocytes, which constitute the majority of cells in the villi of the small intestines. At low glucose concentrations, the predominant pathway is active absorption by Na +/- glucose co-transporter (SGLT1). Intracellular glucose is then assumed to exit across their basolateral membrane via the transporter GLUT2. At some point of glucose concentration, the transport by enterocytes diminishes, upon which a second mechanism enters involving specialized enteroendocrine cells with T1R2/R3 receptors. These cause transient increase of glucose transporter in the enterocytes via intermediate enteric neurons [36-43]. This suggests that any compound that causes a response in oral S fibers will also stimulate these intestinal receptors resulting in increased BG levels and insulin levels, e.g., [42-47]. If the compound does not stimulate T1R2/R3 receptors of the species, such as aspartame on mice [48], it is likely that the sweetener will not facilitate glucose uptake and exert no effect on BG and insulin levels.
It is however, evident that details of these mechanisms are not fully understood. Consequently, a study based on our knowledge of sweet taste in an animal model with and without functioning sweet taste receptors may contribute to the understanding of the role of sweet in intake and control of body weight also in human. The first part of the study will present corroborative data to our previous study on differences in BW, intake and mortality between WT and KO mice [1]. The second part presents data on the relation between BW and blood glucose (BG) and insulin levels. The third part attempts to link gavage of glucose and other sweeteners with effects on BG and insulin levels.
Calhm1 KO have been described cf [16]. Both male and female WT and CALHM1 null mice (KO) were housed individually in the same room and given the same feed (ad lib. Purina 5001). These studies were conducted in accordance to institutional and national guidelines for the care and use of animals and were approved by the University of Minnesota IACUC.
We recorded the relationship between BW and BG and insulin in tail blood of mice of both sexes. For the BG measurements the Alphatrak II glucose meter system was used. According to manufacturers, this device is the only one on the US market certified for BG measurements in mice. We also tried several other systems designed for human blood samples but recorded inconsistent values. Each sample collected 30-50 μl tail blood in a heparinized capillary tube. The tubes were immediately centrifuged, plasma was then transferred to a 0.5 ml micro tube and frozen at -80C in order to be later used for insulin quantification with an ultra sensitive enzyme-linked immunosorbent assay (ELISA) kits from Crystal Chem. Inc. In most cases the analyses were made within one week after blood collection. We strived to use an equal number of WT and KO mice in each experiment to obtain data under the same circumstances.
To avoid excessive gastric distension, food was removed about 4 h before the gavage. We did not measure any consistent difference between BG and insulin levels in samples prior to food removal or 4 h later. Therefore, we call samples taken 4 h after food removal "resting values" and label them "time 0". The first blood collection was followed by gavage, which took generally less than one min. Subsequent samples were taken 20, 40 and 60 min after gavage.
Either 560 mM (10%) glucose, 560 mM (10%) fructose, 10 mM SC 45647 or water were infused with a gastric cannula at a volume corresponding to 1% of the mouse BW. For example, 0.27 ml gavage was administered to a 27 g mouse.
This study began with 29 WT and 23 KO mice. Eighteen months later we have lost 45% of the WT and 26% KO mice. With more than 1/2 of the mice still alive, the final conclusion on rate of mortality has to wait, but our data nonetheless shows a higher mortality in the WT than KO mice.
Our previous study suggests that presence or absence of ability to taste sweet created a difference in intake, which then caused a weight difference between the WT and KO mice. Figure 1a supports this finding by presenting weekly average body weights of 29 WT and 23 KO mice. All mice were fed ad lib. Purina 5001, which contains about 5% fat and approximately 7% carbohydrates with 3.7% sucrose as the major component. Figure 1a shows that the BW difference between the WT and KO mice grew significantly from an average of 1.3 g (WT 21.5 g, SE 0.6 and KO 20.2 g, SE 0.6) to 5 g 21 weeks later (WT 32.5 g, SE 1.4 and KO 27.5 g, SE 0.9).
 Figure 1: Average BW of (A) all WT and KO while fed Purina 5001; (B) WT and KO samples while fed Purina 5015.
View Figure 1
Figure 1: Average BW of (A) all WT and KO while fed Purina 5001; (B) WT and KO samples while fed Purina 5015.
View Figure 1
We assumed that the weight difference was caused by inability of the KO mice to taste sweet and tested this by dividing the mice into 4 groups. We put 10WT and 10 KO mice on Purina 5015 while the remaining two groups of WT and KO continued being fed Purina 5001. Purina 5015 contains more than twice as much fat (11%) as Purina 5001, but considerably less sucrose (0.88%) with lactose (2.7%) as the major carbohydrate. Figure 1b shows that the KO mice on Purina 5015 began putting on weight so after 17 weeks they weighed approximately as much as the WT mice (WT 38.5 g SE 3.8 and KO 37.2 g SE 2.4). During that time the food intake of the KO increased and exceeded that of the WT (489.5 g/week for KO versus 485.0/week g for the WT mice). In the two control groups of 10 mice each kept on Purina 5001, the weight difference was unchanged between WT and KO mice and the KO mice remained lighter than the WT mice.
Figure 2 shows in a 3D plot the relationship of BW on both resting BG and insulin levels in 34 measurements of the WT mice, (BW between 23.1- 43.8 g, average = 31.2 g, open circles) and 32 of the KO mice (BW 21.0 - 43.5 g, average 28.7 g, filled circles). BG is plotted along the X-axis, insulin on the Y-axis and BW on the Z-axis. Each dot symbolizes the intersection of these parameters The correlation coefficients between the three variables BG, insulin and BW in the WT mice varied between r2= 0.61 to 0.66, suggesting a robust relationship. However in the KO mice correlations between the BW/BG was r2= 0.36; BW/insulin r2= 0.71 and BG/insulin r2= 0.30. These will be discussed later.
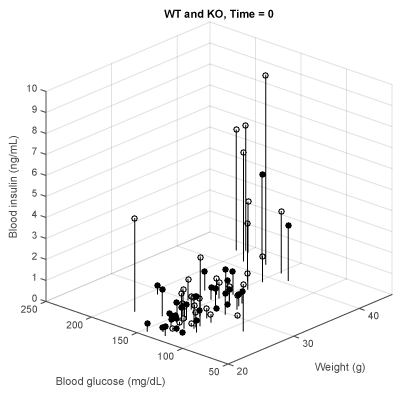 Figure 2: BW vs. BG vs. insulin levels at resting for WT (white, lowest = 23.1 g, mean = 31.2 g, highest = 43.8 g, n = 34) and KO (black, 21.0 g, 28.7 g, 43.5 g, n = 32).
View Figure 2
Figure 2: BW vs. BG vs. insulin levels at resting for WT (white, lowest = 23.1 g, mean = 31.2 g, highest = 43.8 g, n = 34) and KO (black, 21.0 g, 28.7 g, 43.5 g, n = 32).
View Figure 2
We divided the WT and KO mice in a heavy and light group to visualize the influence of BW on BG and insulin levels after glucose gavage. Figure 3a shows BG and insulin levels of heavy mice (open circles) and light (closed circles) of WT mice against time after glucose gavage. The average and range of BW of each group are given in the figure legends. A comparison between heavy and light WT mice shows that the majority of the heavy WT mice combined high BG levels, up to 400 mg/dl, with high insulin response, up to 10 μg/ml, while the BG levels of the lighter mice staid below 300 mg/dl with related lower insulin levels.
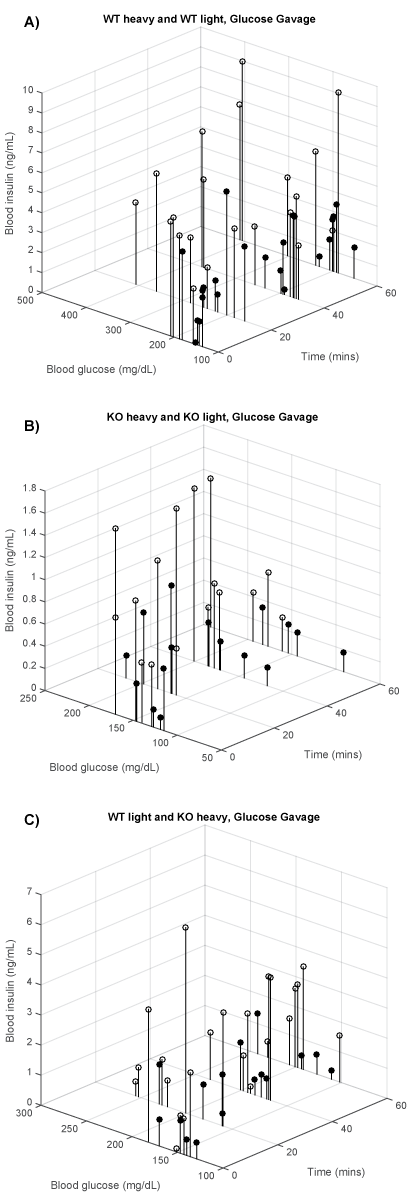 Figure 3: Glucose gavage time vs. BG vs. insulin levels for (A) WT heavy (white, lowest = 37.8 g, mean = 42.2 g, highest = 48.3 g, n = 6) and WT light (black, 24.1 g, 26.6 g, 28.9 g, n = 6); (B) KO heavy (white, 32.7 g, 33.3 g, 34.2 g, n = 4) and KO light (black, 21.0 g, 22.7 g, 24.2 g, n = 4); (C) WT light (white 24.1 g. 34.4 g, 48.3 g and KO heavy (black 21.0 g, 28.0 g, 34.2 g).
View Figure 3
Figure 3: Glucose gavage time vs. BG vs. insulin levels for (A) WT heavy (white, lowest = 37.8 g, mean = 42.2 g, highest = 48.3 g, n = 6) and WT light (black, 24.1 g, 26.6 g, 28.9 g, n = 6); (B) KO heavy (white, 32.7 g, 33.3 g, 34.2 g, n = 4) and KO light (black, 21.0 g, 22.7 g, 24.2 g, n = 4); (C) WT light (white 24.1 g. 34.4 g, 48.3 g and KO heavy (black 21.0 g, 28.0 g, 34.2 g).
View Figure 3
Figure 3b shows BG and insulin levels after glucose gavage in heavy (open circles) and light (closed circles) KO mice plotted against time. A comparison between the location of heavy and light KO groups shows that the majority of the heavy KO mice combined higher BG levels with higher insulin response, around 200 mg/dl and insulin levels up to 1.4 μg/ml, while in the lighter KO mice, BG was less than 200 mg/dl and insulin levels around 0.1 to 0.6 μg/ml.
Figure 3c is included to show in a direct comparison that lighter WT mice had BG and insulin levels exceeding those of heavier KO mice. Data of lighter WT mice (average BW 26.6 g) were plotted against heavy KO mice values (average BW 33.3 g). Thus the KO mice were on the average >6 g heavier than the WT mice. None the less, these lighter WT mice had higher BG and insulin levels than the heavier KO.
Figure 4a displays the BG and insulin levels after gavage with 10% fructose. The 3D plot shows little increase of BG over time and no evident difference between the WT and KO mice. The intercept between BG and insulin at each interval are plotted in figure 4b. Any increase of one or the other parameter should have been seen as drift of the data points to the right and upwards. This is not evident for insulin and small for BG suggesting that gavage of 10% fructose exerted little effects on BG or insulin during 60 min after the gavage.
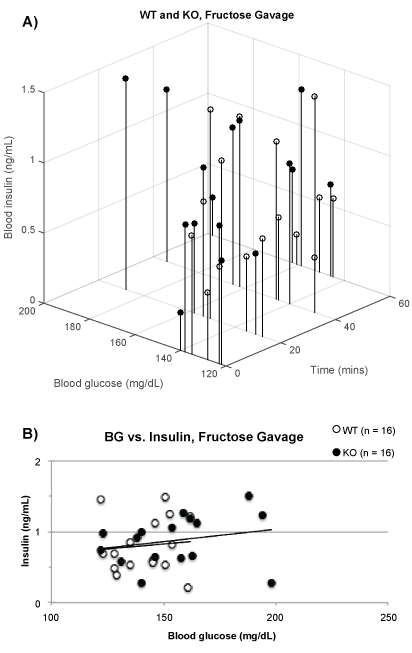 Figure 4: Fructose gavage (A) time vs. BG vs. insulin levels for WT (white, lowest = 24.1 g, mean = 25.4 g, highest = 27.5 g, n = 4) and KO (black, 23.8 g, 27.4 g, 30.3 g, n = 4); (B) BG vs. insulin levels for WT (R2= 0.009) and KO (R2= 0.054).
View Figure 4
Figure 4: Fructose gavage (A) time vs. BG vs. insulin levels for WT (white, lowest = 24.1 g, mean = 25.4 g, highest = 27.5 g, n = 4) and KO (black, 23.8 g, 27.4 g, 30.3 g, n = 4); (B) BG vs. insulin levels for WT (R2= 0.009) and KO (R2= 0.054).
View Figure 4
In figure 5a BG and insulin levels were plotted against time on the Z-axis. It suggests that SC45647 had no effect on BG or insulin of KO (filled circles) mice, but caused some increase of both BG and insulin levels in the WT (open circles) mice.
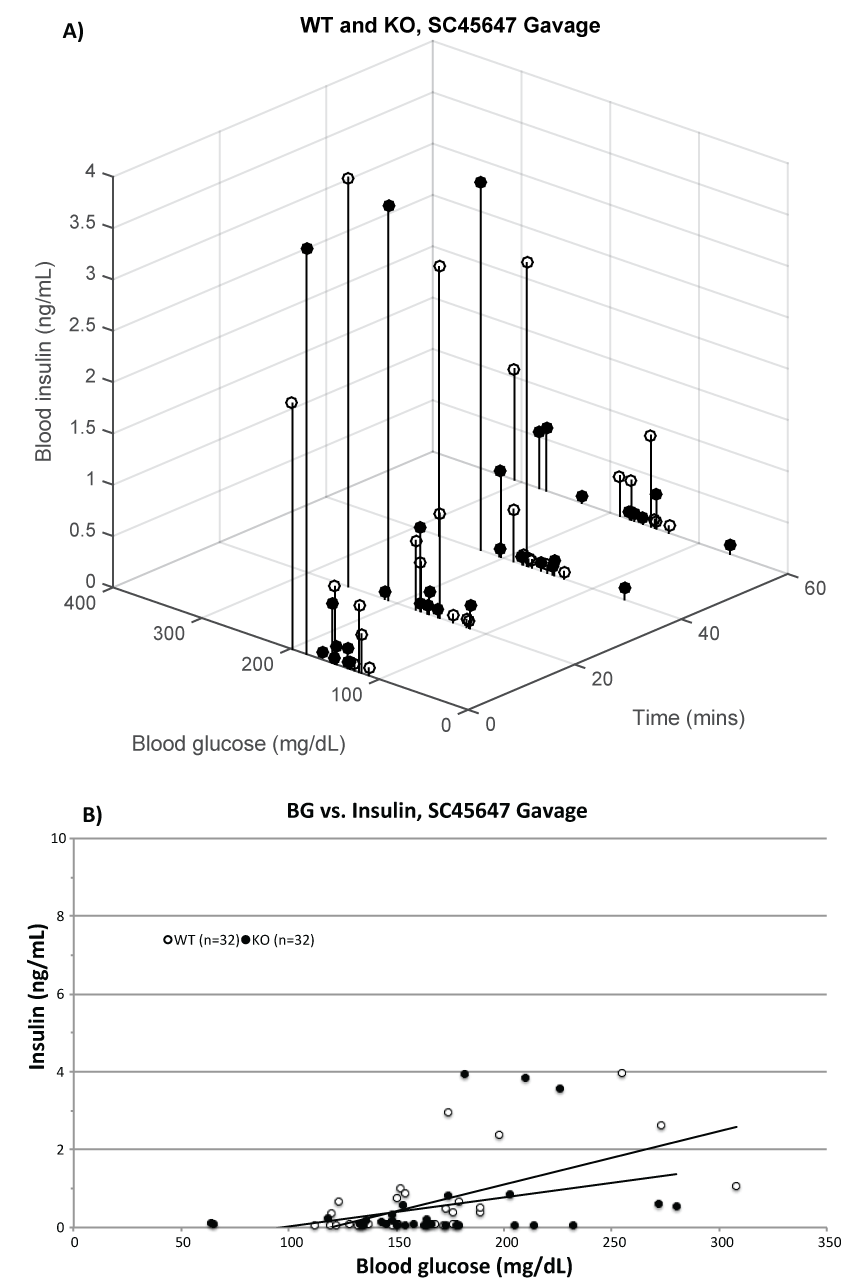 Figure 5: SC45647 gavage (A) time vs. BG vs. insulin levels for WT (white, lowest = 23.1 g, mean = 30.5 g, highest = 43.6 g, n = 8) and KO (black, 22.0 g, 30.1 g, 43.5 g, n = 8); (B) BG vs. insulin levels for WT (R2= 0.381) and KO (R2= 0.103).
View Figure 5
Figure 5: SC45647 gavage (A) time vs. BG vs. insulin levels for WT (white, lowest = 23.1 g, mean = 30.5 g, highest = 43.6 g, n = 8) and KO (black, 22.0 g, 30.1 g, 43.5 g, n = 8); (B) BG vs. insulin levels for WT (R2= 0.381) and KO (R2= 0.103).
View Figure 5
Figure 5b, the diagram plots BG vs. insulin levels for WT (R2= 0.381) and KO (R2= 0.103). The values and the lower regression line suggest that SC45647 caused no change in KO and some increase in WT mice.
Gavage and repeated blood sampling constitute stresses that may affect BG and insulin levels. To test how much these contributed to the findings we report here, we gavaged with water in WT and KO mice. Figure 6 shows that the effect of these manipulations in themselves did not significantly affect our results with sweeteners.
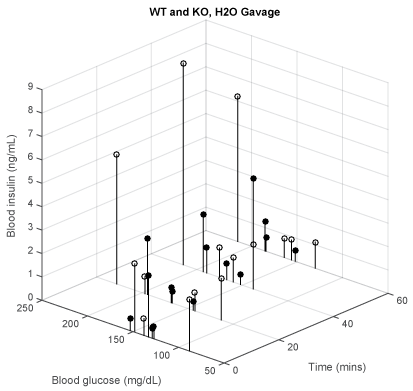 Figure 6: Water gavage- time vs. BG vs. insulin levels for WT (white, lowest = 27.6 g, mean = 32.4 g, highest = 41.7 g, n = 4) and KO (black, 21.4 g, 30.2 g, 40.7 g, n = 4).
View Figure 5
Figure 6: Water gavage- time vs. BG vs. insulin levels for WT (white, lowest = 27.6 g, mean = 32.4 g, highest = 41.7 g, n = 4) and KO (black, 21.4 g, 30.2 g, 40.7 g, n = 4).
View Figure 5
The growth rate of the mice with normal sweet tasting ability (WT) was significantly higher than for the mice with no sweet tasting ability (KO) mice. The result of change of diet to a twice as high fat content made the KO put on weight so that they weighed as much as the WT. Upon return to the normal low fat, higher saccharide diet, the difference in BW between WT and KO returned. This suggests that ability to taste sweet was the cause for the initial weight difference. We recorded a positive correlation between BW and resting BG and insulin levels in both WT and KO mice. But BG and insulin levels were generally less in KO than in WT mice even at the same BW. Both WT and KO mice responded to a load of glucose via gavage with increases of BG and plasma levels, but the rises were smaller in KO mice. Fructose elicited minimal increases of BG or insulin levels. SC45647 had a small effect on BG and insulin levels in WT but in KO mice virtually no effect.
We will first relate body weight, food intake and mortality in WT with functioning intestinal T1R2/R3 receptors and KO that lack functioning oral receptors with our previous study [1]. The second part deals with the relationship between body weight (BW) and resting blood glucose (BG) level in KO and WT mice. We will then discuss the effect of glucose, fructose and SC 45647 gavage on BG and insulin level of WT and KO.
As mentioned all mice were fed Purina 5001 with 5% fat and approximately 7% saccharides with 3.7% sucrose as the major sweetener when the difference in BW shown in figure 1a developed. We attribute the heavier BW of the WT to the sweet taste of sucrose, which stimulated their intake, but not in the KO mice that could not taste its sweetness. Consequently they did not eat more than their needs of energy demanded. We tested this conclusion by increasing the fat content and lowered saccharides.
The more than double (11%) fat content of Purina 5015 improved the food palatability for the KO mice in figure 1b. The cause for this is either post-a factors from the digestive system or oral fatty acid receptors, such as GPR40 in Type I TRCs, which should be unaffected to the deletion of CALHM1 [49]. The latter explanation is also supported by our own TBP tests showing that the KO mice can discriminate between water and 3% oil emulsion. This conclusion does not exclude post-ingestive factors from the digestive system. In contrast, to the WT mice Purina 5015, containing considerably less carbohydrate ( < 3%) and 0.88% sucrose, was less attractive than 5001. Consequently they ate less than the lighter KO mice.
The important point here is, however, that the KO mice were capable to reach the same BW as the WT mice and that they did not weigh less because of loss of CALHM1 functions. The most likely explanation is absence of ability to taste sweet by the KO mice. This conclusion is also supported by data in mice that do not taste sweet because of deletion of TRPM5 channels in their Type II TRCs [50].
In regard to lower mortality of the KO, we have at this point no definitive data corroborating our previous study [1] since many are still alive, but our data to date suggest similar results. Furthermore, corroborative data have been published from both rodents and primates, restriction of food was the cause for the increased life spans [51-53]. The difference between these earlier studies and this one is that the animals were ad-lib-fed animals that limited their intake.
Generally it is considered that obesity is associated with increased risk of developing insulin resistance and type 2 diabetes e.g., [8,54]. In humans BG after glucose gavage returns faster to pre gavage BG levels in subjects with lower BW [55]. Here the correlation coefficients between BW and BG were the highest in the WT mice (r2= 0.61). The WT mice are also the animal model most representative of the human situation, because of their functioning T1 receptors and greater ease to become over weight. The positive correlation between BW and BG in KO was much less (r2= 0.36) suggesting that with blocked intestinal R2/R3 receptors increased BW would not necessarily raise BG levels. Furthermore, KO mice with the same BW as WT mice show a slower rise in both BG and insulin levels with increasing BW than WT mice. Similar relationship has been reported in monkeys where "Fasting basal insulin and glucose concentrations are lower in DR (dietary restricted) compared to control animals while insulin sensitivity is higher in the restricted animals" [51]. Applied to the human situation, this suggests that blocking intestinal R2/R3 receptors would not only lead to lower BG but also lower insulin levels.
It is well established that T1R2 and T1R3 receptors are present in entero-endocrine cells of the small intestines were they facilitate [42], uptake and transports of glucose and fructose into the blood [43,56].
As noted in figure 3a and figure 3b, glucose gavage increased BG and insulin response less in the KO mice with deficient T1R2/R3 receptors than in WT mice with functioning receptors. Figure 3c demonstrates this further in a comparison of BG and insulin levels in a group of lighter WT mice and heavier KO mice. In spite of the latter's significantly larger BW their BG and insulin levels were less. This suggests that T1R2/R3 deficiency lowered glucose adsorption. It lowered glucose adsorption. It is possible that the BG and insulin difference between WT and KO mice reflects the contribution of intestinal T1R2 and R3 receptors to glucose adsorption.
Finally, since insulin increase stimulates intake in both mice and humans, the results here take the effects of eating less and live longer one step further, by demonstrating that diminishing input from Type II TRCs with T1R2/R3 receptors lowers not only food intake but also lowers glucose adsorption from small intestines thereby reducing BG levels and insulin levels enabling glucose homeostasis [57].
Figure 4a and figure 4b show a difference between carbohydrate metabolism of glucose and fructose. As opposed to isomerizing into glucose, its reducing sugar counterpart, our data demonstrate that fructose more readily converts to fructose-1-phosphate and instead enters metabolism as a glycolytic intermediate. Thus, no BG change directly stems from the gavage. The absence of an effect on BG or insulin in figure 4a and figure 4b corroborates this conclusion.
Non-saccharide sweeteners provide sweetness to foods and beverages without adding calories. However their usefulness in weight reduction has recently been questioned? It is possible based on the notion that their sweet taste stimulates oral and intestinal T1 R2/R3 receptors, with enhanced glucose adsorption and increased BG, which then increases hunger and stimulates food intake, thereby causing weight gain cf [7].
With gavage we avoided the cephalic or oral phase of a sweetener that is one minor factor in insulin release. However, figure 5a shows that BG and insulin levels were slightly increased in the WT mice and figure 5b suggests a BG increase after gavage with SC45647. These increases were much smaller than after glucose gavage.
To humans 10 mM SC45647 is significantly sweeter than 10% glucose. In WT mice 10 mM SC45647 is strongly preferred and elicits a much larger taste nerve response than 10% glucose [16]. If the small effects on insulin levels in WT mice also applies to humans, it suggests that non-saccharide sweeteners exert less effects on BG and insulin than saccharides even if they taste sweeter. In the KO, the effect of SC45647 on BG and insulin levels did not differ from that of water, because KO mice don't taste and show no taste nerve response to SC45647 [16].
As mentioned, gavage and repeated blood sampling constitute stresses that may affect BG and insulin levels. Therefore, we recorded the effects of gavage with water in WT and KO mice. The results in figure 6 suggest that these manipulations could not by themselves be the cause for the increase of BG or insulin levels that are reported here.
The molecular structure of a taste receptor will determine if a compound is able to stimulate its TRC because the TRC and its taste fibers determine the taste quality of the stimulus. Compounds that stimulate oral Type II TRCs with T1R2/R3 receptors trigger impulses in synapsing nerve fibers that give rise to a taste quality that causes intake. In humans these nerve impulses is perceived as pleasant, stimulates intake also in the innate and classified as sweet by the adult. This is a fundamental principle of taste coding from the periphery, whether it is on the tongue or in the intestines [58,59]. Blocking oral T1R2/R3 receptors removes the S fiber portion from the taste fiber input. This diminishes the palatability of most food and lowers the consumption and reduces greatly the temptation of over consumption. Intestinal TRCs with T1R2/R3 receptors increase significantly intestinal glucose adsorption. This will affect BG and insulin levels. Blocking intestinal T1R2 or R3 receptors decreases glucose uptake and lowers BG and insulin levels. Effects on the GLUT2 and SGLT1 mechanisms on intestinal glucose adsorption can explain our data on intake, choice of food, BG and insulin in WT and KO mice. In humans a continuous 5 to 10% decrease of intestinal adsorption could over time bring BW to a healthy level of an over-weight or obese individual. If the block is limited to intestinal sweet receptors, the effect should be subconscious, which probably do guarantee that it will not affect intake.
All authors declare there is no conflict of interest.