Astroblastomas are rare tumours of the central nervous system, and only a few cases of brain stem/primary spinal cord astroblastoma have been reported. It has been suggested that EWSR1/BEND2 fusion defines an epigenetically distinct subtype of astroblastoma. We present a rare case of primary intramedullary spinal cord astroblastoma with EWSR1/BEND2 fusion. To our knowledge, this is only the third reported case in the literature with this specific diagnosis, the first in an adult female, and the first that has been treated upfront with total surgical resection.
Spine, Spinal cord, Neoplasms, Neuroepithelial, Molecular biology, Pathology, Neurosurgery
Astroblastomas are rare tumours of the central nervous system with a predilection for young females [1]. They are defined by the 2021 WHO classification as a distinct entity - Astroblastoma MN-1 altered, within the category of circumscribed astrocytic gliomas [2]. The first case of spinal cord astroblastoma was only reported in 2018 [3]. Only a few cases of primary brain stem/spinal cord astroblastoma have been reported since, and it has been suggested that EWSR1/BEND2 fusion defines an epigenetically distinct subtype of astroblastoma [4].
We present a rare case of primary intramedullary spinal cord astroblastoma with EWSR1/BEND2 fusion. To our knowledge, this is only the third reported case in the literature with this specific diagnosis, the first in an adult female, and the first that has been treated upfront with total surgical resection.
A 45-year-old female with no significant background medical history presented to the outpatients department of our institution with a six month history of progressive myelopathy. Her symptoms began with tingling in the fingers bilaterally, which slowly progressed to reduced dexterity and clumsiness. On examination, tone and power were intact in the muscle groups of all four limbs. She was hyper-reflexic throughout, with bilateral Hoffmann’s sign, Babinski’s sign, and clonus. Sensation was reduced in the fingers and the feet bilaterally and not in any particular dermatomal distribution, but proprioception was preserved. She had difficulty finger tapping. Her gait was normal.
MRI imaging of her cervical spine (Figure 1) revealed an intramedullary lesion from C4 to C6, which was relatively well demarcated post-Gadolinium enhancement, with the radiological differential diagnosis including an ependymoma or astrocytoma. There was also significant associated oedema within the cervical spinal cord. Given the imaging findings and clinical progression, surgery was performed.
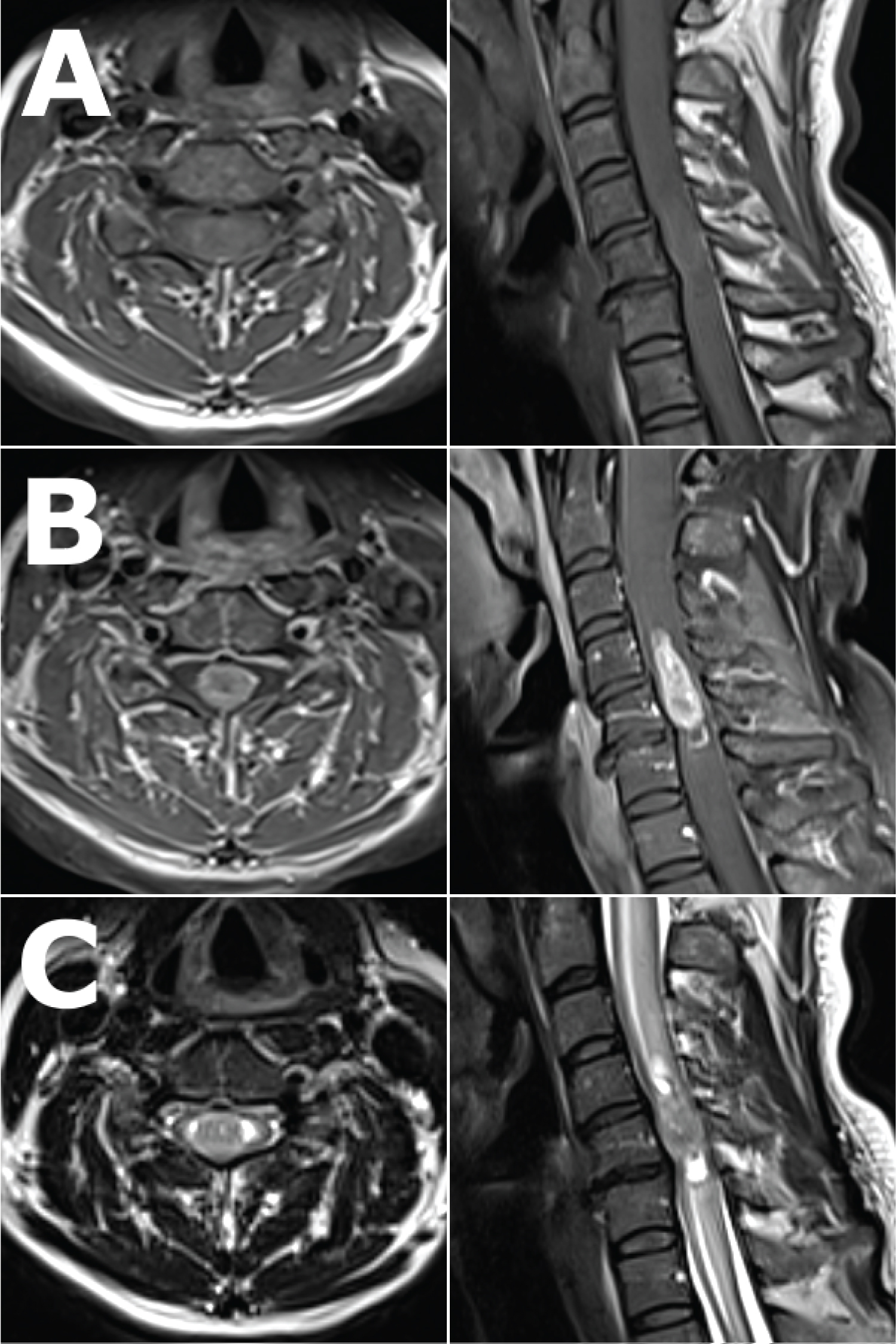 Figure 1: Pre-operative axial (left) and sagittal (right) views of the cervical spine MRI showing the lesion. T1 weighted images, A) pre and B) post- Gadolinium enhancement, showing a well-defined, T1 isointense but homogenously enhancing intramedullary lesion from C4 to C6, occupying most of the spinal canal at the level of C5. C) T2 weighted images showing the same lesion, which is T2 isointense with tiny spotty T2 hyperintensities within, splaying the normal spinal cord circumferentially around it at the level of C5, with associated syringomyelia most evident at the superior and inferior ends of the lesion, and with marked associated spinal cord oedema as evidenced by T2 signal change from C1 to T1.
View Figure 1
Figure 1: Pre-operative axial (left) and sagittal (right) views of the cervical spine MRI showing the lesion. T1 weighted images, A) pre and B) post- Gadolinium enhancement, showing a well-defined, T1 isointense but homogenously enhancing intramedullary lesion from C4 to C6, occupying most of the spinal canal at the level of C5. C) T2 weighted images showing the same lesion, which is T2 isointense with tiny spotty T2 hyperintensities within, splaying the normal spinal cord circumferentially around it at the level of C5, with associated syringomyelia most evident at the superior and inferior ends of the lesion, and with marked associated spinal cord oedema as evidenced by T2 signal change from C1 to T1.
View Figure 1
A posterior approach was taken and C3 to C6 laminectomies performed. The spinal cord was visibly under pressure following durotomy. Intraoperative neurophysiological monitoring was employed and a midline myelotomy was performed with the assistance of dorsal column mapping. A soft pale grey and tan lesion was encountered which was relatively avascular. The intraoperative frozen section analysis suggested an ependymoma. A clear plane between the lesion and normal spinal cord tissue could not be reliably identified but given the frozen section diagnosis, gross total resection of the lesion was performed. Motor evoked potentials were reduced in the right upper limb and somatosensory evoked potentials were reduced globally by the end of the procedure.
Neuropathologic examination of the tumour demonstrated features consistent with an astroblastoma (Figure 2). Following DNA methylation profiling and Next Generation Sequencing, the final diagnosis was that of a neuroepithelial tumour with features of astroblastoma, EWSR1/BEND2 fusion positive. The myelopathic symptoms of the patient were worse in the immediate post-operative period. She suffered no focal power deficit but she had new and severe neuropathic pain and spasticity in her hands. Post-operative imaging confirmed a radiological total resection of the lesion, with no recurrence at 18 month follow up, and she has received adjuvant radiation and Temozolomide.
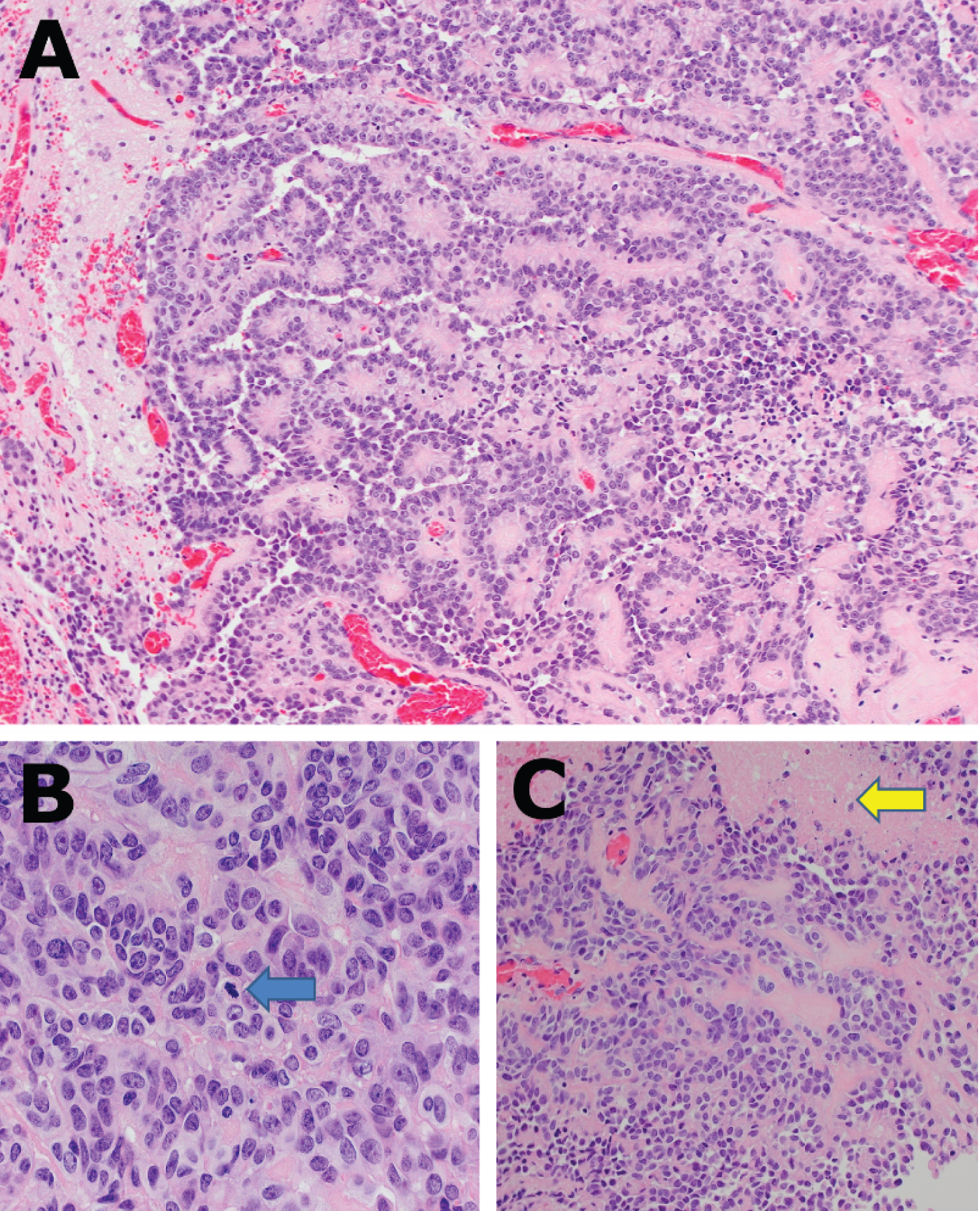 Figure 2: A) Cellular neoplasm composed of oval to columnar cells with hyperchromatic apically placed nuclei, variably prominent central nucleoli and a moderate amount of eosinophilic cytoplasm are arranged in papillary structures and perivascular pseudorosettes. H&E x20; B) Frequent mitoses: 3-5 per 10 high power fields (blue arrow) H&E x40; C) Multifocal coagulative necrosis (yellow arrow), H&E x20.
View Figure 2
Figure 2: A) Cellular neoplasm composed of oval to columnar cells with hyperchromatic apically placed nuclei, variably prominent central nucleoli and a moderate amount of eosinophilic cytoplasm are arranged in papillary structures and perivascular pseudorosettes. H&E x20; B) Frequent mitoses: 3-5 per 10 high power fields (blue arrow) H&E x40; C) Multifocal coagulative necrosis (yellow arrow), H&E x20.
View Figure 2
Prior to 2021, astroblastomas were diagnosed purely on histological features showing tumour cells in characteristic perivascular pseudorosettes [5]. Astroblastoma is now defined by the latest WHO classification as harbouring an alteration in the MN1 gene, typically MN1/BEND2 fusion [6]. Recent reports including the current study have shown astroblastoma-like tumours with EWSR1/BEND2 gene fusion without MN1 abnormalities suggesting an epigenetically distinct subtype of astroblastoma. Another group reports astroblastoma with MAMLD1/BEND2 gene fusion, histologically indistinct from the previously described subgroups [7]. These tumours appear to have a predilection for brainstem and spinal cord; however the clinical significance of these putative astroblastoma subgroups remains unknown.
Astroblastoma was once thought to be confined to the brain with some reports of metastases to the spine [8,9]. Primary astroblastoma of the brainstem and spinal cord has only recently been described [3,4,10-12]. Patients present with myelopathy or focal neurological deficit corresponding to the spinal level of disease [4]. The imaging appearances are fairly consistent - T1 weighted MRI shows a homogenously enhancing well circumscribed lesion, and T2 weighted MRI may show small cysts within a T2 iso to slightly hyperintense lesion with associated spinal cord oedema [3,4,10-12]. Progression of disease ranges from slow over months such as in the current case to very quickly over days to weeks [11]. Watchful waiting has been trialled but ultimately all cases required surgery and adjuvant treatment due to clinical and radiological progression of disease [10].
All known cases of brainstem and spinal cord astroblastoma have had surgery, however the extent of resection varied from biopsy only [3,4], partial debulking [11,12], subtotal resection [10], and total resection in the current study. It has been described as grey in colour and with no clear plane identifiable intraoperatively in keeping with the observations of the current study [3,12]. Radical tumour resection is the most effective treatment for cerebral astroblastoma [13], however previous reports of surgical treatment of brainstem and spinal cord astroblastoma have been limited by fear of causing irreversible neurological deficit due to the lack of a surgical plane [3].
Adjuvant chemotherapy includes the use of Temozolomide, Bevacimumab, and Etoposide, and adjuvant radiotherapy has also been used [10-12]. The outcome from the disease is difficult to predict due to its rarity, but half of patients with this disease have died, and one of the two known survivors has widespread dissemination of disease at last follow up. A clinical summary of known cases of primary intramedullary brainstem & spinal cord astroblastoma with EWSR1/BEND2 fusion can be found in Table 1.
Table 1: Clinical summary of known cases of primary intramedullary brainstem & spinal cord astroblastoma with EWSR1/BEND2 fusion. View Table 1
In summary, we present a rare case of primary intramedullary spinal cord astroblastoma with EWSR1/BEND2 fusion treated upfront with complete surgical excision. We focus on the clinical and surgical descriptions of the disease. More case reports are required to understand the natural history and response to treatment of this extremely rare disease.
Not applicable.
Not applicable.
The authors would like to thank the department of neuropathology at Great Ormond Street Hospital, London, the United Kingdom, for their assistance in performing Next Generation Sequencing to make the molecular diagnosis.