We report a case of a segmental neurofibromatosis or type V neurofibromatosis, a rare genetic disorder defined by a limitation of multiple macular cutaneous pigmentations “cafe-au-lait” (CAL) spots and/or neurofibromas to a single, unilateral segment of the body. A 34-year-old woman presented to our Deparment with tender, subcutaneous, palpabale nodules arising from previous episiotomy scar 2 years postpartum. Physical examination revealed similar nodules with same morphological characteristics in the left labia majora and the right ingvinum. Histopathological analysis of the nodule revealed a neurofibroma. The cells were immunohistochemically CD34 and S-100 positive. On the left shoulder “cafe-au-lait” macules (CALMs) as a skin hyperpigmentation were seen. The disease may have malignant alteration, thus regular follow up examinations should be considered.
Segmental neurofibromatosis, Type V neurofibromatosis, Episiotomy scar
Neurofibromatosis (NF) are a group of genetic disorders that primarily affect neuroectoderm [1]. It is a common disorder affecting 1 from 3000 persons [2]. Since it is extremely variable in its presentation, Riccardi suggested in 1982, a useful system for classifying neurofibromatosis into eight types [3]. One of these types, segmental neurofibromatosis (NF-V) is defined by a limitation of multiple macular cutaneous pigmentations “cafe-au-lait” (CAL) spots and/or neurofibromas to a single, unilateral segment of the body. Distribution of this spots is mostly on the trunk, head and neck [2,3]. Postzygotic somatic mutation, in a primitive neural crest cell, is the most likely causative mechanism, and thus lesions should be strictly unilateral and noninherited [1,2]. It is very rare with an incidence 20 times lower than neurofibromatosis [2].
A 34-year-old woman gravida 1, para 1, was admitted to the Gynecology Department with a inicially discomfort, dull ache in the perineum postpartum, but over the past 2 years it had increased to a sharp, focal pain over the left aspect of the perineum. The patient was able to palpate a tender mass arising from previous episiotomy scar. Physical examination was remarkable for a 1 cm tender, mobile nodule in the subcutaneous tissue just to the left of the midline of the perineum. Similar nodules with same morphological characteristics have been palpated in the left labia majora and the right ingvinum. A routine gynecologic examination revealed fullness in the right ingvinum, whilst the size of the uterus was physiological for age, in retroverted position. A transvaginal ultrasound revealed a conglomerate of enlarged nodules, the largest 25 mm in diameter just beneath the left ovary. On the left shoulder, “cafe-au-lait” macules (CALMs) as a skin hyperpigmentation 3 cm in diameter were seen. Patients medical history was unremarkable. A Pap smear was negative for intra-epithelial lesions and malignancy. Magnetic resonance imaging revealed presacral multiple rounded hyperintense lesions 35 mm in diameter with hypointense centrum. The same consistency lesions were found in the subcutaneus pelvic tissue, mainly glutealy and close to vagina 22 mm in diameter. Some of the lesions were seen in the sacral canal and sacral foramina, 20 mm in diameter. Biopsy of the subcutaneous nodule was taken and revealed a neurofibroma. Results of laboratory evaluation and of tumor markers were within the reference values (AFP 1.8 ug/L, CEA 0.7 ug/L, CA 125 31.0 kIU/L, CA 19-9 11.3 kIU/L and CA 15-3 6.4 kIU/L).
Gross examination of the resected subcutaneous nodule demonstrated a gelatinous lesion measuring 1.1 cm in diameter, arising in previous episiotomy scar. Histopathological analysis revealed interlacing bundles of elongated cells with wavy, darkly stained nuclei. No mitotic figures were seen. Stroma contained variable mucin and interspersed variably sized collagen bundles. Diagnosis of a neurofibroma was made based on analysis of the sample. The cells of this component obtained with ultrasound guided puncture of the nodule were immunohistochemically CD34 and S-100 positive. No immunolabeling was disclosed for SMA and Caldesmon. Immunohistochemical staining of B-cathenin was positive in cytoplasm.
Neurofibromatosis are a complex genetical disorder involving cells derived from the neural crest. Tremendous clinical variability is seen. Eight types of the disorder were established by Riccardi in 1982 [2].
Neurofibromatosis type I (von Recklinghausen disease, NF-1) is an autosomal dominant disease characterized primarily by CALMs, Lisch nodules (pigmented hamartomas in the iris) and peripheral neurofibromas (especially optic nerve gliomas and astrocytomas), kyphoscoliosis, pseudoarthrosis, pheochromocytoma and medullary carcinoma of the thyroid gland. Begins early in the childhood with CALMs. Most of the adults develop neurofibromas, benign tumors that are usually located on or just under the skin. Thus some of them are at risk of transformation to malignant peripheral nerve sheath tumors [1,3,4].
Neurofibromatosis type II (NF-2) is an autosomal dominant disease, central neurofibromatosis characterised by bilateral vestibular schwannomas or acoustic neuromas. Begins in adolescence with loss of hearing, tinnitus and balance problems [3].
Segmental neurofibromatosis or neurofibromatosis type V (NF-5) is a rare form of neurofibromatosis first introduced as a concept in 1956. by Crowe, et al. [2] Its prevalence is between 0.0025-0.002% and occurs twice as more often in females [2,3]. It is an example of mosaicism in which localized disease results from a postzygotic NF1 gene mutation located on the proximal long arm of chromosome 17. Gene mutation can arise in both somatic and gonadal cell lines. Gonadal mosaicism is thought to be responsible for reports of patients with localized disease having children with generalized NF-1 [3,5]. The phenotype of the disease is linked with the somatic NF1 gene mutation, the type of it, the number of the cells affected by it and the region of the body. Although there is a low amount of affected cells, next-generation sequencing (NGS) is a diagnostic tool option of early diagnosis [6]. Peaks bimodally, at 10-30 years and 50-70 years [2,3]. It is characterized by multiple macular cutaneous pigmentations, “cafe-au-lait” spots that can be absent or limited to one dermatome as well as neurofibromas limited to one or adjacent nerve roots. Tumors usually grow slowly, are mostly asymptomatic and range in size from 0.1 cm to several centimeters in diameter. They tend to arise in dermatomal distribution following the lines of Blaschko, most commonly in cervical, followed by thoracic, lumbar, and sacral region. Some patients have had complications of NF-1, which have included learning difficulties, optic pathway gliomas, and pseudoarthrosis [2,3]. Those with learning difficulties tended to have larger areas of cutaneous involvement. Lisch nodules were rarely seen. Malignancies associated with are mostly ones derived fom neural crest cells, peripheral nerve sheath tumors and malignant melanomas [2,7,8] (Figure 1, Figure 2 and Figure 3).
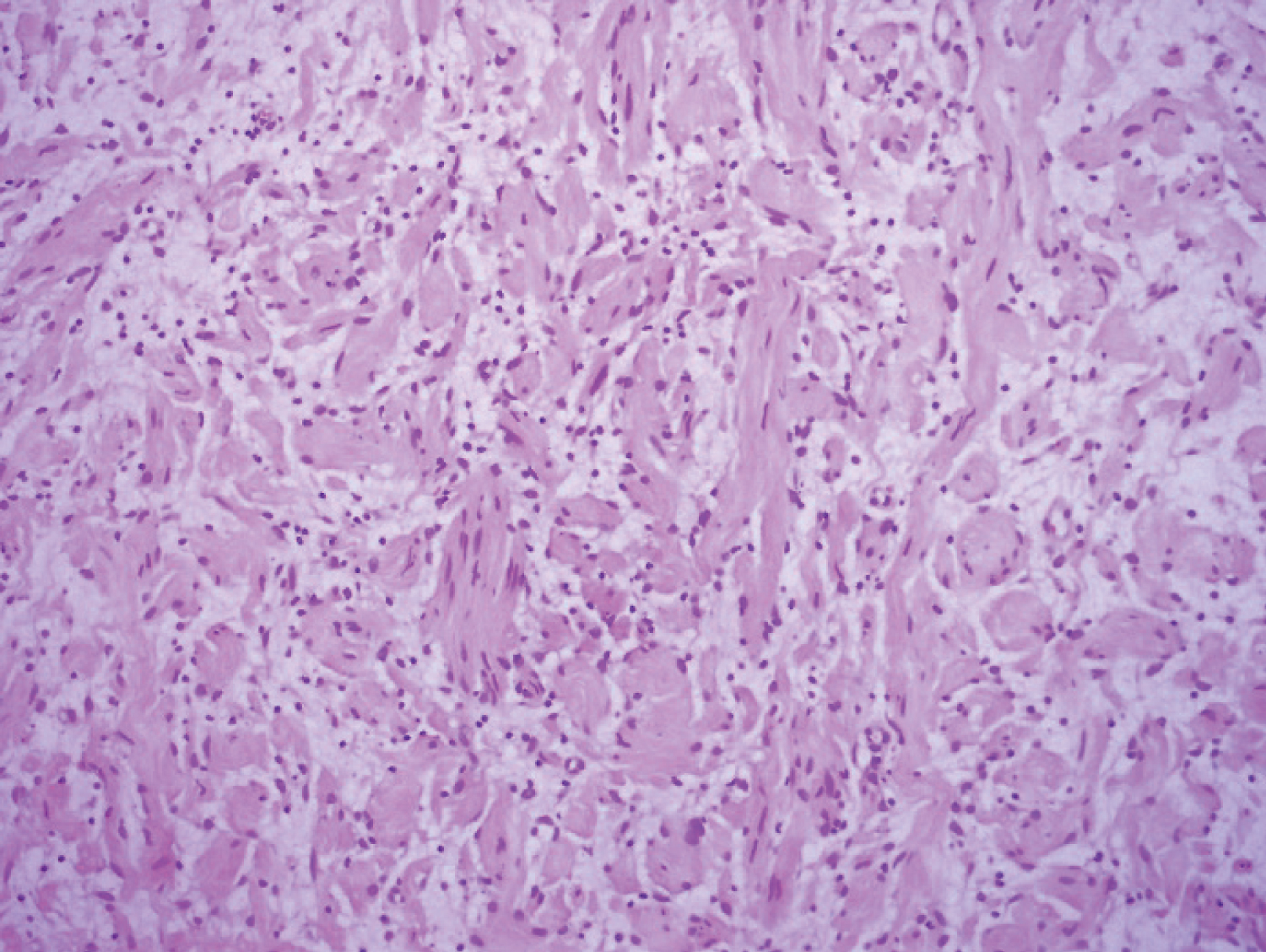 Figure 1: Histological aspects of segmental neurofibromatosis (hematoxylin and eosin staining).
View Figure 1
Figure 1: Histological aspects of segmental neurofibromatosis (hematoxylin and eosin staining).
View Figure 1
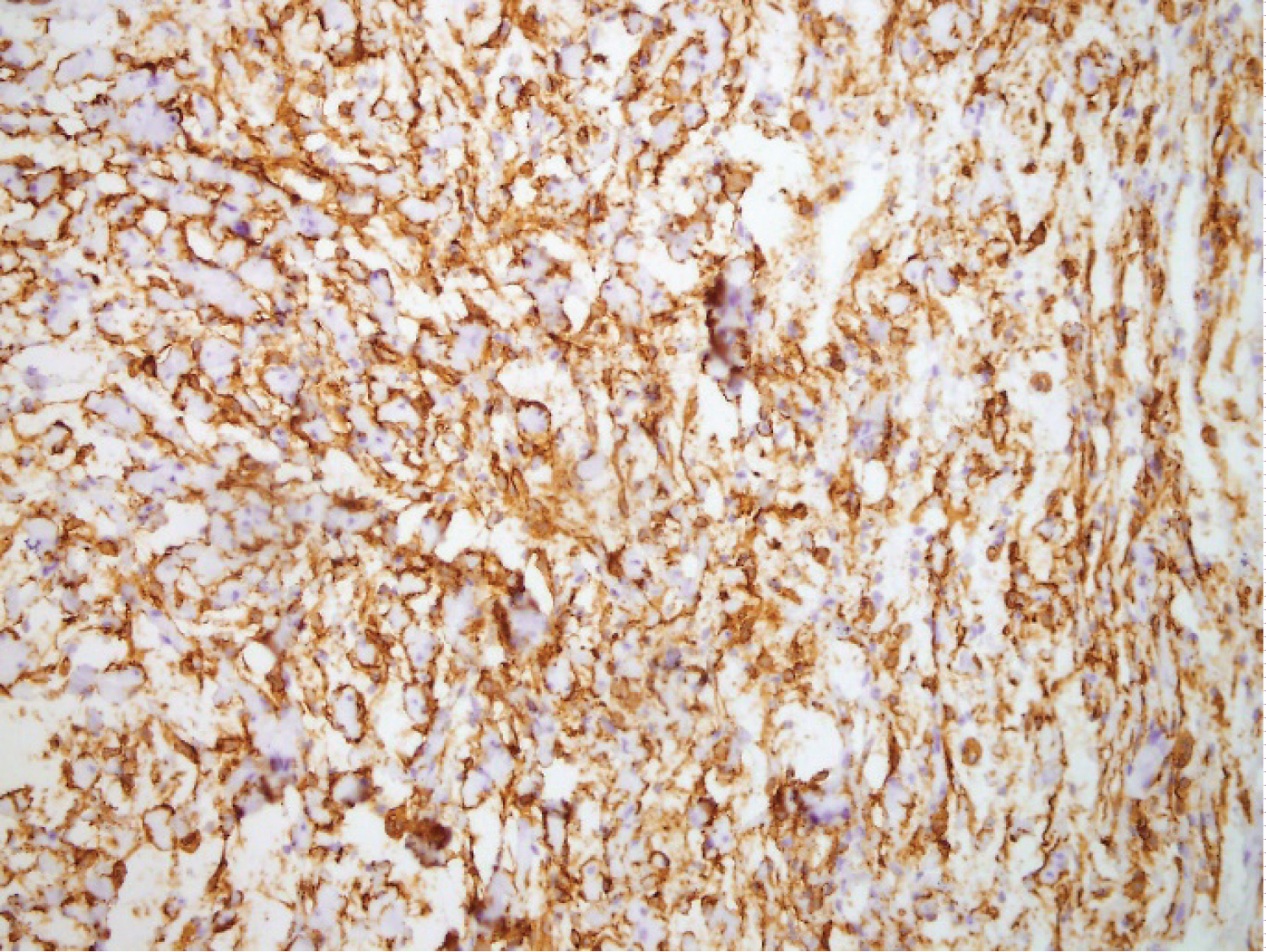 Figure 2: Intense positivity for CD34 protein in segmental neurofibromatosis.
View Figure 2
Figure 2: Intense positivity for CD34 protein in segmental neurofibromatosis.
View Figure 2
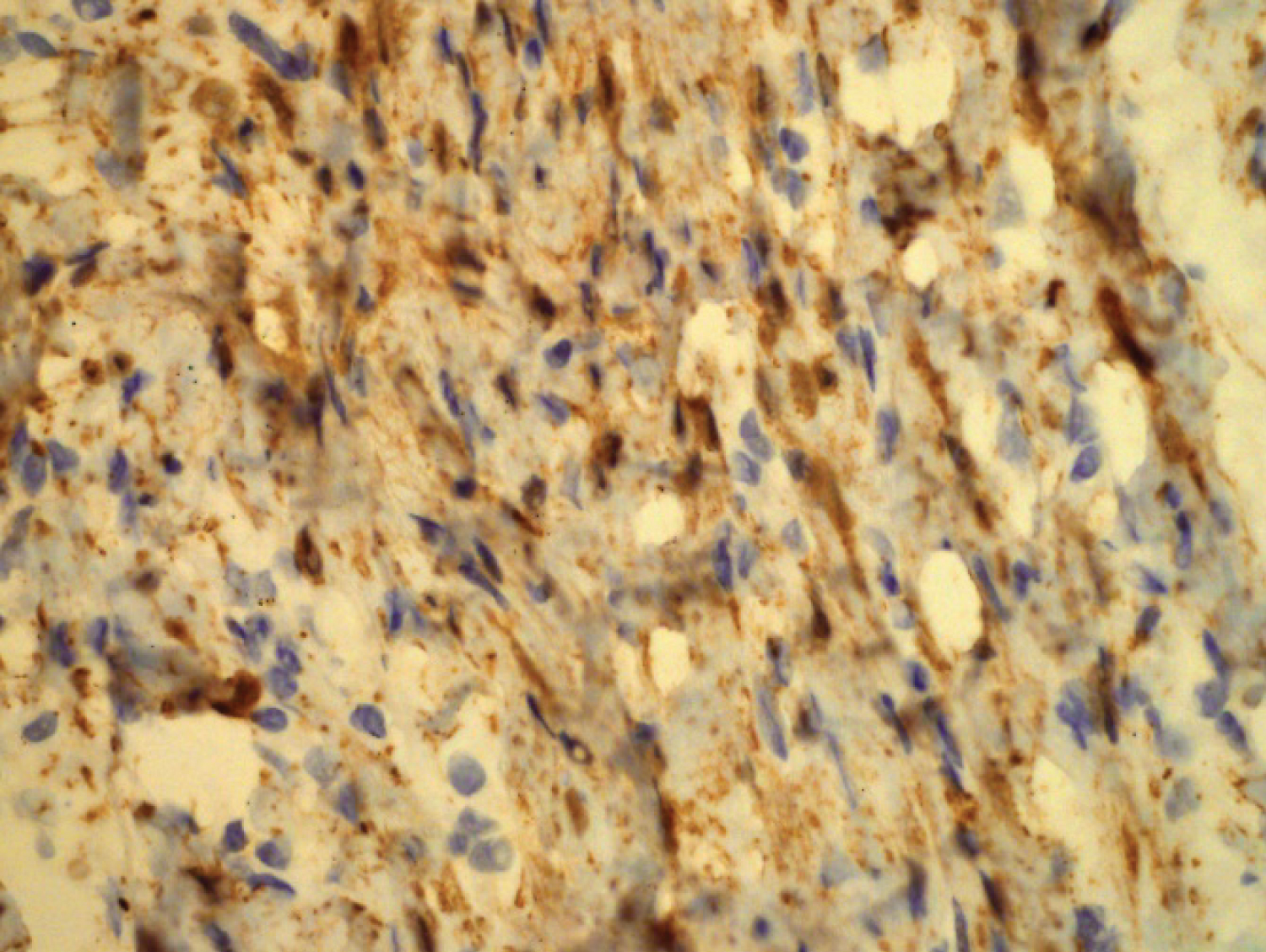 Figure 3: Intense positivity for S100 protein in segmental neurofibromatosis.
View Figure 3
Figure 3: Intense positivity for S100 protein in segmental neurofibromatosis.
View Figure 3
There are no specific guidelines regarding management of segmental neurofibromatosis. Patient should be informed that they do not have generalized NF-1 and their risk of disease associated complications is low.
None.
None.
The authors declare that they equally contributed regarding the publication of this paper.