Homocysteine is an amino acid with an SH group, metabolised by the remethylation and transsulfuration pathways. Several genetic and environmental factors (like deficient nutrition status, systemic disease or consumption of certain drugs), can lead to changes in the levels of plasma homocysteine.
Nowadays, hyperhomocysteinemia is considered an important and independent risk factor for atherosclerosis and cardiovascular disease.
Several pathological mechanisms have been proposed for the effect of hyperhomocysteinemia in the development of cardiovascular disease. Among them are DNA methylation, decreased protein S-nitrosylation, production of reactive oxidative species and decrease in nitric oxide formation.
Main strategies being tested for the treatment of this condition involve supplementation of folic acid, vitamins B6, B12 or riboflavin. From these, increased plasma folic acid levels by folate-rich diet or pharmacological supplementation seems to be the most effective.
Homocysteine, Cardiovascular disease, Folic acid, Vitamin B6, Vitamin B12, Riboflavin
Cardiovascular diseases are the main cause of death in developed countries. Several risk factors are involved in its development like family background, hypercholesterolemia, obesity, diabetes mellitus and environmental factors such as sedentarism. High level of plasma homocysteine also represents a risk factor, independent of other factors. It is observed that 5-7% of general population show hyperhomocysteinemia, and of those, of the ones having atherosclerotic vascular disease, 13-47% show moderate to intermediate hyperhomocysteine levels [1,2].
Changes in plasma homocysteine levels can result from the presence of several factors including physiological, genetic, nutritional, drug induced and hormonal factors. First studies indicating mild hyperhomocysteinemia as an independent risk factor for cardiovascular disease, with a prevalence of approximately 5% in the general population, date from the 80th decade [3-6].
Homocysteine is an amino acid with SH group, produced exclusively from methionine which, as an essential amino acid, comes from food diet [7]. Homocysteine metabolism involves two different metabolic pathways: the remethylation and the transsulfuration pathways (Figure 1) [8,9].
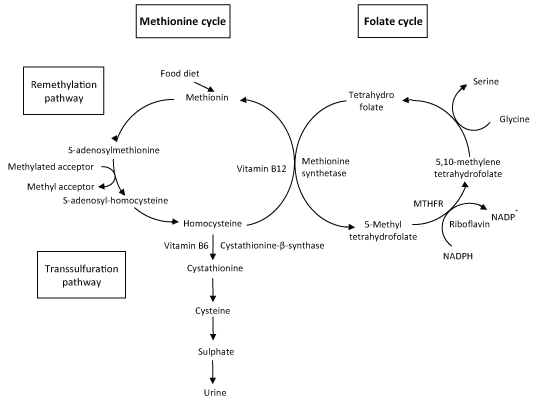 Figure 1: Homocysteine metabolism (adapted from [10]).
View Figure 1
Figure 1: Homocysteine metabolism (adapted from [10]).
View Figure 1
The remethylation pathway, mainly occurring in starvation conditions, involves the catabolism of methionine. In the first step, methionine reacts with ATP leading to the formation of S-adenosylmethionine. This molecule then loses the methyl group attached with the sulphur atom (present in methionine), forming S-adenosyl-homocysteine. The methyl group released by S-adenosylmethionine can be transferred by methyl-transferases to several substrates (methyl acceptors) like proteins, DNA or phospholipids (Figure 1).
The next step is the hydrolysis of S-adenosyl-homocysteine to produce homocysteine. Later, the sulphur atom of homocysteine can be transferred to serine entering the transsulfuration pathway described below. Homocysteine can also be used to regenerate methionine, by the vitamin B12 dependent enzyme methionine synthetase (MS) that transfers a methyl group given by 5-methyltetrahydrofolate to homocysteine. In most cells, this is the main or unique pathway of conversion of homocysteine to methionine. This amino acid will enter a new cycle of donation of methyl group. Another product of this reaction is tetrahydrofolate, that will be reconverted to 5-methyltetrahydrofolate through the folate cycle, where the conversion of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate is catalysed by the FAD dependent enzyme, methylenetetrahydrofolate reductase (MTHFR) [10]. MTHFR enzyme has an important function since it regulates the availability of 5-methyltetrahydrofolate for homocysteine remethylation. The reaction catalysed by MS allows the connection between the methionine and the folate cycles (Figure 1).
The transsulfuration pathway, that takes place in cases of methionine overload, involves the catabolism of homocysteine to sulphate, later excreted in the urine. The sulphur atom present in homocysteine is transferred to serine leading to the production of cysteine, while the nitrogenated group and the carbons that belonged to homocysteine are released as ammonium and α-ketobutirate. In this process, two enzymes are sequentially involved: Cystathionine β-synthase (CBS) and cystathionine lyase. The active form of vitamin B6 - pyridoxal phosphate - is a cofactor of CBS that converts homocysteine into cystathionine, first step of the transsulfuration pathway (Figure 1). The α-ketobutirate can be converted into propionyl-CoA that is further transformed into succinyl-CoA (an intermediate of the Krebs cycle). It is evident the importance of B vitamins in homocysteine metabolism, since these molecules play a crucial role as enzyme cofactors. Vitamin B6 is required for CBS activity while vitamin B12 is the cofactor of MS. Another important vitamin for homocysteine metabolism is riboflavin that is necessary for MTHFR function.
Homocysteine can be found in plasma in the free form (around 20-30%), but most of it (70-80%) is associated with plasma proteins, mainly albumin.
Plasma homocysteine present in the free form can also be in the oxidized form, giving rise to two types of disulphides: Homocysteine dimers (homocystine) or homocysteine-cysteine dimers. From the free homocysteine fraction, 2 to 5% correspond to its reduced form. Total plasma homocysteine is the sum of all free forms and forms attached to proteins [11].
Concerning plasma homocysteine levels, there is no consensus about the upper standard limits for healthy individuals, ranging from 5 to 15 μmol/L [2,12,13]. However, some studies have shown that an upper limit of 15 μmol/L is too high in well-nourished populations without vitamin deficiencies [14]. Moreover, plasma homocysteine concentrations vary with age and gender [12,13].
It has been documented the existence of substantial cardiovascular risk with plasma homocysteine levels between 10 and 15 μmol/L. However, on the other hand, de Bree, et al. [15] observed that an increase of 5 μmol/L in plasma homocysteine concentration, rises 1.03 the risk of coronary heart disease mortality. These results do not support the idea of an influence of hyperhomocysteinemia in increased risk of cardiovascular disease [15].
The clinical syndrome homocystinuria, identified almost 60 years ago [16,17], is a disorder with autosomal recessive transmission. Phenotypic characteristics include thromboembolism and vascular occlusion, changes in long bones, ocular displacement, mental retardation and varying degrees of neurological impairment [10,18,19].
Severe hyperhomocysteinemia is caused by rare genetic defects in the remethylation or transsulfuration pathways. The most common inborn error of homocysteine metabolism is the CBS deficiency, also called classical hyperhomocysteinemia, that affects the transsulfuration pathway. Thirty-three point mutations in the long arm of chromosome 21 (21q22.3) responsible for this enzymatic deficiency have been described [20]. The incidence of homozygous individuals for the CBS mutation in the world is 1:200.000, while for heterozygous varies between 1:70 and 1:2000 in the general population [21]. According with Tsai, et al. [11], it is estimated that 30-40% of individuals with early vascular disease, are heterozygous for the CBS mutation.
So far, more than 150 different mutations in the CBS gene have been identified in different populations [22]. Examples of these mutations are nine distinct mutations identified by Mendes, et al. [23] in 22 independent alleles from homocystinuria patients: Four novel mutations (p.K269del, p.P427L, p.S500L and p.L540Q) and five previously described mutations (p.P49L, p.C165Rfs*2, p.I278T, p.R336H and p.D444N). Another study identified two novel missense mutations (p.L136P and p.A158V), in the CBS gene of a Han Chinese family [22].
Two novel CBS mutations were identified in work of Ibrahim, et al. [24], a missense change in exon 7 (p.L156P) and an in-frame deletion (c.808_810del; p.E270del) in exon 10. These researchers also identified a recurrent missense mutation (p.T257M) in exon 10 of the gene [24].
Li, et al. [25] detected eleven mutations and eight (IVS3+1G>A, p.T493fsX46, p.T236N, p.L230Q, p.K72I, p.S201ProfsX36, p.M337IfsX115 and IVS14-1G>C) were novel. The remaining three mutations (p.R125Q, p.T257M and p.G116R) had been previously reported [25].
Moreover, Voskoboeva, et al. [26] detected one new nonsense mutation (Q368Term), one new missense mutation (р.D444Y) and three novel small deletions (c.1560-1569delCACCGGGAAG; c.216-217delAT; c.1498-1499delT) in Russian patients.
Other rare genetic changes can cause an up to 20 times increase in plasma homocysteine and lead to the development of severe hyperhomocysteinemia and result from deficiency in the MTHFR enzyme of the remethylation pathway and 10 different mutations in this gene have already been described (located at 1p36.3) [18,19].
The major known genetic determinant of mild hyperhomocysteinemia in the general population is a thermolabile missense mutation, that leads to the change of alanine per valine, resulting from the substitution of cytosine per thymine in base 677 of the gene. This mutation leads to loss of more than 60% of MTHFR enzymatic activity [20], and 8% of general population is homozygous for this variant. In these patients, folate level interferes directly with plasma homocysteine content, and this is only increased when folate concentration is below 15.4 nmol/L [27-30]. This mutation can be present together with the CBS mutation.
The other rare genetic change results from mutation in the MS gene, being associated with ischemic cardiac disease, mainly in smokers [20].
In most cases, severe hyperhomocysteinemia develops in patients homozygous for the CBS mutation and in only in 5-10% of individuals deficient in MTHFR. Heterozygous individuals for CBS and MTHFR deficiencies, when associated with low level of folate and use of drugs develop moderate or intermediate hyperhomocysteinemia [18].
Mutations in the genes MTHFR and MS lead to clinical symptoms like the ones described for the CBS mutations, suggesting that the presence of high levels of homocysteine might be the pathogenic factor involved in vascular complications [19].
Although innate errors of homocysteine metabolism previously described are very rare, hyperhomocysteinemia is relatively common in several populations. It is more common in men (21% above women) and increases in women during menopause [28]. There is also an increase with age and, since elderly patients show bigger vitamin deficiency, hyperhomocysteinemia is, in this group of individuals, an important risk factor for coronary disease [29,30].
Several studies discuss the effect of external factors in the development of high levels of plasma homocysteine. One of these factors are nutritional changes. Nygard, et al. [31] describe a strong inverse association between plasma concentration of folic acid and/or food diet and total concentration of plasma homocysteine [31]. Folic acid deficiency can lead to increased plasma homocysteine [31]. Individuals with folate levels above 15 nmol/L do not show hyperhomocysteinemia [31].
Moreover, according with the study of Fonseca, et al. [7], patients with vitamin B6 deficiency, have normal basal homocysteine but altered levels after overload. However, other studies observed that folate, vitamin B6 or B12 insufficiencies were associated with hyperhomocysteinemia in both sexes [32]. On the other hand, deficiency of vitamin B2 was significantly associated with hyperhomocysteinemia only in males. In individuals with adequate folate, vitamins B6 and B12 levels, there was no significant association between vitamin B2 and hyperhomocysteinemia [32]. The association was only observed in individuals who had poor folate, vitamin B6, or B12 status [32]. Same researchers also observed that the association between vitamin B12 insufficiency and hyperhomocysteinemia was not affected by simultaneous vitamin B2 or B6 insufficiency but increased 3-fold when combined with folate deficiency [32].
These results suggest a synergic relationship of folate and vitamin B12 deficiencies on the development of hyperhomocysteinemia. The same happens with folate and vitamin B6 insufficiencies [32].
The use of certain drugs can also indirectly lead to increase in plasma homocysteine. During therapy with methotrexate (structural analogue of folate) and anticonvulsants (phenytoin and carbamazepine), folic acid levels are decreased [18] and can result in increase in homocysteine levels. This statement is consistent with work of Chandrasekaran, et al. [33] where a correlation between the duration of antiepileptic drug therapy and high homocysteine plasma levels was observed. However, a meta-analysis performed to determine the influence of oxcarbazepine monotherapy on plasma homocysteine levels, does not support the hypothesis that the therapy changes serum levels of homocysteine [34].
Moreover, the use of the anaesthetic nitrous oxide reduces vitamin B12 levels and vitamin B6 is altered by theophylline, consequently affecting homocysteine content [18].
Hormonal changes, like oestrogen decrease during menopause, increases homocysteine levels. Thyroid hormones also interfere with plasma homocysteine. It has been observed that patients with hypothyroidism show high homocysteine levels, while hyperthyroidism leads to normal levels [7].
Other work explored the contribution of nitrative stress on the deficiency of CBS activity [35]. This study observed that an increased nitrative stress level was accompanied by increased plasma homocysteine. Moreover, increased nitration of CBS led to decreased CBS activity [35].
Moreover, Yakub, et al. [36] observed a relationship between lead and homocysteine plasma levels. These researchers found out that individuals with genotype CT or TT for the MTHFR gene who also had blood lead levels above 10 μg/dL, had significantly increased risk for hyperhomocysteinemia when compared with the CC genotype [36].
Studies have also been conducted on the importance of microbial infection in homocysteine levels and consequent atherogenesis. Pathogenic microorganisms cause synthesis of polyamines in host cells. This is accomplished by increasing the transfer of aminopropyl groups from adenosylmethionine to putrescine, that leads to depletion of intracellular adenosylmethionine in those cells. Absence of adenosylmethionine dysregulates methionine metabolism, leading to hyperhomocysteinemia, among other effects [37,38].
Hyperhomocysteinemia has been considered an important risk factor, independent of other factors, for cardiovascular disease and atherosclerosis [39,40].
So far, there is no consensus concerning the pathophysiological mechanism explaining the relationship between hyperhomocysteinemia and cardiovascular disease, and several hypotheses have been suggested. In this way, vascular lesion resulting from hyperhomocysteinemia, would involve lesion of endothelial cells, growth of vascular smooth muscles, increase in platelet adhesion and oxidation of LDL followed by its deposition in the vascular wall and the activation of blood coagulation cascade [39,41].
Studies have also shown that high levels of homocysteine have impact on the traditional cardiovascular risk factors. For instance, hyperhomocysteinemia seems to influence plasma cholesterol levels. Choy, et al. [42,43] observed that high levels of homocysteine lead to increased production of cholesterol and secretion of apolipoprotein B-100. This effect on cholesterol synthesis is mediated by activation of the enzyme HMG-CoA reductase, that catalyses the regulatory point in cholesterol biosynthesis [42,43]. Wu, et al. [44] also observed a markedly increase in HMG-CoA reductase activity in liver cells of hyperhomocysteinemic rats, together with hepatic lipid accumulation. Activation of HMG-CoA reductase was caused by increased gene expression and reduction in enzyme phosphorylation (that converts it in the inactive form) [44]. According to Choy, et al. [42] the increase in apoB secretion was caused by the increase in cholesterol level induced by homocysteine, being a plausible mechanism for the observed relationship between hyperhomocysteinemia and the development of atherogenesis and coronary artery disease.
Moreover, Hemati, et al. [45] observed that insulin resistant patients usually have higher homocysteine plasma level. This study allowed establishing a relationship between high homocysteine levels and high plasma glucose observed in diabetes mellitus type 2 patients [45].
Other studies have associated hyperhomocysteinemia with increased blood pressure [46,47]. However, work of Borges, et al. [48] does not corroborate the possible role of homocysteine on changes in blood pressure.
In vivo studies performed by Dudman [49] showed that homocysteine activates separately each type of leucocyte and endothelial cell, giving evidences of the role of homocysteine as a natural regulator of leucocytes. Leucocyte induction and endothelial adhesion provoked by homocysteine, leads to trans-endothelial migration of leucocytes and endothelial lesion. Consequently, selective change of the expression of the protein responsible for chemoattraction of monocytes and interleukins will take place. These lead to the release of cytokines and inflammatory agonists [49].
Study of Poddar, et al. [50] showed that homocysteine induced mRNA expression of the proinflammatory cytokines monocyte chemoattractant protein-1 and interleukin-8 in cultured human aortic endothelial cells, and their secretion. This suggests that homocysteine may contribute to the initiation and progression of vascular disease by promoting leukocyte recruitment [50].
Moreover, Silverman, et al. [51] observed that hyperhomocysteinemia leads to a 5-fold increase in vascular cell adhesion molecule 1 (VCAM-1) mRNA expression in human endothelial cells, resulting in increased adhesion of monocytes.
Fang, et al. [52] demonstrated that hyperhomocysteinemia and hyperglycemia increased monocytes and macrophages lesions, followed by increased levels of inflammatory monocytes and macrophages and consequent accelerates atherosclerosis. Inflammatory monocytes correlated negatively with the ratio of S-adenosylmethionine to S-adenosyl-homocysteine [52].
An alternative explanation for the effect of hyperhomocysteinemia is that it is a marker of low level of vitamins B or diminished capacity of methylation in cells, being any of these two factors possibly related with the disease. Castro, et al. [53] showed that leucocytes of patients with cardiovascular disease have decreased DNA methylation, together with increased plasma levels of homocysteine and S-adenosyl-homocysteine. Moreover, even with a general state of hypomethylation, certain regions of the genome can be hypermethylated.
Studies have suggested that a better biomarker of the association of hyperhomocysteinemia and vascular disease would be the increase in S-adenosyl-homocysteine, instead of homocysteine itself [54,55]. The enhancement of S-adenosyl-homocysteine results from the reversal reaction catalysed by S-adenosyl-homocysteine hydrolase. The increase in S-adenosyl-homocysteine leads to feedback inhibition of methyltransferases that are dependent on S-adenosylmethionine, like DNA methyl-transferases [54,55]. Therefore, there will be a decrease in DNA methylation. Results of Castro, et al. [53] support the hypothesis of S-adenosyl-homocysteine accumulation and consequent DNA hypomethylation are the causing agents of the pathogenic role of hyperhomocysteinemia in vascular disease. These disturbances in DNA methylation and consequent changes in gene expression, have deep effects in the risk of cardiovascular disease [56].
In the same way, Zhang, et al. [57] observed that mild increased homocysteine levels induce hypomethylation, while high concentration of homocysteine lead to hypermethylation in the promoter CpG island of the dimethylarginine dimethylaminohydrolase (DDAH2) gene. An increase in DDAH2 gene expression was observed in mild enhanced concentration and in high concentration of homocysteine a decrease in gene expression was detected [57]. This protein is the key enzyme for degradation of asymmetric dimethylarginine (ADMA), which is an endogenous inhibitor of endothelial nitric oxide (NO) synthase (eNOS). The inhibition of DDAH2 activity was followed by increase in ADMA and consequent reduction of eNOS activity and decrease of NO production [57]. Dysfunction of nitric oxide pathway, resulting from hyperhomocysteinemia, that leads to reduced dilator responses of arteries and arterioles was proposed as one of the pathomechanisms that would affect all cell functions involved in cardiac dysfunction [58].
Yan, et al. [59] also showed that hyperhomocysteinemia reduces NO levels, phosphorylation of endothelial nitric oxide synthase (eNOS) and vascular endothelial growth factor expression, leading to endothelial dysfunction.
Moreover, increased S-adenosyl-homocysteine leads, not only to DNA hypomethylation, but also to decreased methylation of tRNA [60], as well as to decreased protein arginine methylation [61]. Esse, et al. [61] state that protein arginine methylation is more sensitive to increases in S-adenosyl-homocysteine than DNA methylation.
Changes in tRNA methylation lead to altered expression of the selenoprotein glutathione peroxidase 1, one of the major antioxidants that can modulate overall oxidative stress [60]. The decrease in its gene expression leads to an increase in cellular hydrogen peroxide and consequent up-regulation of endothelial adhesion molecule expression. This is enough to provoke changes in cell adhesion by increasing the binding of leukocytes. This study demonstrates that an increase in S-adenosyl-homocysteine can result in oxidative stress [60].
Moreover, recent studies have suggested that alterations in hydrogen sulfide (H2S) production may also contribute to homocysteine associated vascular disease. H2S was found to be a gasotransmitter, like nitric oxide and carbon monoxide [62]. Production of H2S results from the activity of several enzymes, including CBS [63]. An alternative reaction catalysed by CBS is the condensation of cysteine with homocysteine to form cystathionine and H2S [64,65]. Chen, et al. [65] have shown that at physiologically relevant concentrations of serine, homocysteine, and cysteine, about 5% of the cystathionine formed is from cysteine. S-adenosylmethionine stimulates H2S producing reaction [65]. H2S is produced in endothelium, participating in the regulation of endothelial integrity through its beneficial anti-inflammatory and antioxidant effects. Hence, hyperhomocysteinemia is usually associated with decreased levels of H2S and this may contribute to the development of atherosclerosis.
Another proposed mechanism is the simultaneous increase of the activity of thromboxane A2 (TXA2) both in vessels and platelets [58]. Koller, et al. [58] suggested that these changes result from an increased production of reactive oxidative species (oxidative stress) due to increased NADPH oxidase assembly, which eventually leads to an inflammatory process and morphological remodelling of vessels.
In cases of hyperhomocysteinemia, the increase in superoxide production by NADPH oxidase and reduced formation of NO alters the regulation of mitochondrial function in the myocardium [58,66].
According with Chen, et al. [67] severe oxidative stress may lead to cell death through apoptosis or necrosis. Besides NADPH oxidase, there are other stimuli for the production of reactive oxidative species in cells that include mitochondria where, under oxidative stress, the free electrons on the mitochondrial electron transport chain may leak out and react with molecular oxygen, thereby generating superoxide anion as metabolic by-products during respiration. Moreover, NO may react with superoxide anions producing the highly reactive peroxynitrite anion that damage DNA, proteins and lipids. These alterations in macromolecules play an important role in many physiological and pathological conditions [67].
A recent study found that hyperhomocysteinemia caused mitochondrial ultrastructural injury, translocation of cytochrome c and inhibited the activities of complexes I, II and III from the electron transport chain [68]. These researchers also observed an increase in reactive oxidative species levels together with enhanced mitochondrial pSTAT3, a transcription factor that regulates expression of many other genes involved in cell proliferation, differentiation and survival [68]. This study concludes that high homocysteine levels cause mitochondrial dysfunction, which was displayed as a significant decrease in respiratory chain activities, increased production of reactive oxidative species and mitochondrial swelling [68].
Zhang, et al. [69] also observed, together with decreased activity of complex III, low levels of aconitase gene expression (enzyme involved in the Krebs cycle) in hyperhomocysteinemia conditions. Increased levels of reactive oxidative species and decreased ATP content, that play important roles in cellular stress response during cell growth, were also present [69]. Hyperhomocysteinemia induced mitochondrial dysfunction that modulated apoptosis [70], leading to coronary and peripheral artery and venous vessel diseases, resulting in vasomotor dysfunction and increased thrombosis, consequently increasing morbidity and mortality [58].
Oxidative stress seems to be an important in vivo mechanism responsible for the development of endothelium dysregulation of wall shear stress in hyperhomocysteinemia cases [58,71].
Increased oxidative species, decreased endothelial nitric oxide synthase and consequent reduced nitric oxide levels were accompanied by reduced levels of protein S-nitrosylation in cases of hyperhomocysteinemia [72-74]. Results of these studies suggest that homocysteine promotes atherosclerosis by inhibiting vascular protein S-nitrosylation [72-74].
As previously discussed, infections by pathogenic microbes leads to depletion of adenosylmethionine that causes, besides hyperhomocysteinemia, a decrease in the production of nitric oxide and impaired host response to infectious microbes, contributing to the pathogenesis of dementia and atherosclerosis [37,38].
All mentioned changes, that result from hyperhomocysteinemia, impair endothelium-dependent vasodilation and regulation of blood flow, apoptosis of endothelial cell that can be an important mechanism of vascular injury, resulting in vascular leak, inflammation and coagulation, leading to the development of atherosclerosis and cardiovascular disease [75].
As previously described, 5-methyltetrahydrofolate is the substrate for the enzyme methionine synthetase, being produced from folic acid. A strong inverse association between folate concentration and plasma homocysteine has been described by Nygard, et al. [31].
One approach tested in several studies for the treatment of hyperhomocysteinemia involves supplementation of folic acid. The study Homocysteine Lowering Trialist Collaboration [76] showed that supplements of 0.5-5.0 mg/day of folic acid, lead to a decrease of 25% in plasma homocysteine.
On the other hand, a study of Wald, et al. [77] in patients with cardiovascular disease, allowed to establish a food supplement of 0.8 mg/day to reach maximal drop of plasma homocysteine. Tighe, et al. [78] observed that a maximum homocysteine decrease in patients with cardiac disease, can be obtained with folic acid supplements like the ideal dose for healthy individuals.
Malinow, et al. [1] observed a reduction of plasma homocysteine levels by using breakfast cereals fortified with folic acid in patients with coronary heart disease. These researchers showed that plasma homocysteine decreased proportionally with the increase in plasma content in folic acid and this one was affected by the folic acid content of the breakfast cereal [1]. Cereals providing 127 μg/day of folic acid, increased plasma folic acid by 31%, but decreased plasma homocysteine by only 3.7%. But cereals giving 499 and 665 μg/day of folic acid, increased plasma folic acid by 64.8% and 105.7%, respectively, and decreased plasma homocysteine by 11% and 14%, respectively [1].
Fabre, et al. [79] also obtained a 32.3% reduction in plasma homocysteine by oral supplementation of 5 mg/day of levofolinic acid to healthy women aged 18-35 years for 30 days. A decrease in plasma homocysteine was observed from the second day of treatment onwards and maximum decline was seen after 30 days [79].
However, Armada, et al. [80] tested the supplementation of folic acid or folinic acid to haemodialysis patients and no normalization of homocysteine levels was observed.
A recent study of Tian, et al. [81] tested the efficacy of folic acid therapy in patients having hyperhomocysteinemia. After 3 months of treatment, plasma homocysteine levels of 484 patients were reduced to below the normal levels (15 μmol/L), corresponding to an average reduction of 56% [81]. However, these researchers observed that more than 40% of patients subjected to folic acid supplementation failed to reach the normal homocysteine range (5-15 μmol/L) after 3 months of treatment [81].
Zappacosta, et al. [82] tested the efficacy of a folate-rich diet or pharmacological supplementation of individuals with moderate hyperhomocysteinemia, on plasma homocysteine decrease. They observed that supplementation with natural folate-rich foods, folic acid or 5-methyltetrahydrofolate allowed a similar reduction in homocysteine levels in blood [82].
Similar results were obtained by Chandrasekaran, et al. [33] with supplementation of folic acid to hyperhomocysteinemic children receiving phenytoin and carbamazepine monotherapy. These researchers observed that the duration of antiepileptic drug therapy correlated significantly with high homocysteine and low folic acid plasma levels and administration of folic acid reduced plasma homocysteine content [33].
Other studies show that vitamin B12, important cofactor of the enzyme methionine synthetase, has lower effect in the decrease of plasma homocysteine, when compared with folic acid [76]. In this study, supplements of vitamin B12 (0.5 mg/day) allowed an additional decrease of 7% in plasma homocysteine [76]. Quilivan, et al. [83] observed that when folate levels are optimized, a dependence between vitamin B12 and plasma homocysteine develops, that is not observed during folic acid supplementation.
Azadibakhsh, et al. [84] tested a combined treatment of folic acid and vitamin B12 and concluded that oral supplementation with 15 mg/day folic acid together with 1 mg/day of vitamin B12 is effective in reducing homocysteine levels in haemodialysis patients.
Supplements of pyridoxal phosphate, active form of vitamin B6 (cofactor of the enzyme cystathionine β-synthetase) do not have any additional effect to folic acid and vitamin B12 supplementation [76]. However, work of McKinley, et al. [85] with healthy individuals between 63-80 years old, showed that supplement of low dose of vitamin B6 (1.6 mg/day) for 12 weeks, after folic acid supplementation (that leads to 19.6% homocysteine reduction), leads to a subsequent decrease of 7.5% in plasma homocysteine levels.
No consensus is observed among the results obtained in several studies with B vitamin supplementations. However, according with Debreceni and Debreceni [86,87] the failure of vitamin therapies may result from inappropriate designs, since most studies neglected the impact of the use of other treatments, like statins, acetylsalicylic acid and other drugs, concomitant with B vitamin supplementations. These researchers state that the effect of statins and aspirin on cardiovascular disease protection and prevention might have reduced or abolished the possibility of observing an effect of B vitamin supplementation. In this way, further research is required to evaluate the real effect of vitamin substitution therapy on decreasing homocysteine plasma levels and cardiovascular disease prevention [86,87].
Riboflavin supplements (required for the MTHFR enzyme) are not effective in plasma homocysteine reduction, before or after optimization of folate levels [88]. However, homozygous individuals for the C677T mutation on the MTHFR gene show riboflavin dependence [89,90].
Moreover, Moat, et al. [91] measured plasma homocysteine in 126 healthy individuals 20-63 years of age with different MTHFR genotypes (42 CC, 42 CT, and 42 TT) at baseline and after three supplements for 4 months: Placebo plus natural diet; 400 μg/day of folic acid supplement plus natural diet; and increased dietary folate to 400 μg/day. This study showed that at baseline and after nutritional intervention, lower riboflavin status was associated with increased plasma homocysteine concentrations. Moreover, at baseline, individuals with the TT genotype had significantly higher homocysteine levels than individuals with the CT and CC genotypes [91]. Plasma folate was lower in the TT group than in the CT or CC genotypes. CT individuals had intermediate levels [91]. Results of this study suggest that an increase in folic acid intake might also increase the requirement for riboflavin to achieve maximal catalytic activity of the enzyme MTHFR. However, no association between riboflavin status and vascular endothelial function at baseline or after increased intake from fortified foods was observed, suggesting that the increase in riboflavin intake may not have additional benefits to the cardiovascular system [91].
A long-term treatment with vitamin C has been applied by Bagi, et al. [71] to rats having hyperhomocysteinemia to prevent oxidative stress-induced dysregulation of arteriolar wall shear stress observed in these cases. Rats subjected to this treatment showed reduced oxidative stress and increase in nitric oxide bioavailability. The regulation of shear stress in arterioles was restored and systemic blood pressure normalized [71].
Other strategies have been tested for the treatment of hyperhomocysteinemia like the administration of cadmium in hyperhomocysteinemic mice, since this condition may result in susceptibility to xenobiotics like cadmium. In the study of Ramambason, et al. [92] no difference in plasma homocysteine level was found after cadmium administration in control and hyperhomocysteinemic mice. However, exposed hyperhomocysteinemic mice showed significant lower levels of glutathione when compared to control mice, reflecting oxidative stress [92].
A recent study of Haddadi-Guemghar, et al. [93] used prune extract, a good source of phenolic antioxidants, to investigate its effect on hyperhomocysteinemic male mice. These researchers observed that the administration of prune extract significantly decreased plasma homocysteine level [93].
Kondakçi, et al. [94] tested the effect of N-acetylcysteine on plasma homocysteine levels and observed a decrease in serum homocysteine together with reduction of hepatic and renal reactive oxygen species. Studies of Chen, et al. [65] provide a biochemical explanation for the homocysteine-lowering effects of N-acetylcysteine treatment. Decreased H2S levels are found to be pathogenic in human disease. In this way, it may be possible to increase H2S and lower homocysteine levels by increasing the concentration of cysteine in tissues. N-acetylcysteine is converted to cysteine inside cells, thereby increasing the concentration of cysteine and thus driving the conversion of homocysteine to cystathionine. This treatment would also increase the production of H2S [65].
Moreover, another study evaluated the effect of quercetin administration on homocysteine metabolism and hepatic antioxidant status in rats fed with high methionine diet [95]. This study reports that plasma homocysteine increased after high methionine diet and quercetin supplementation was able to lower those levels. However, this effect was accompanied by a decrease in serum and liver glutathione content and increased plasma alanine aminotransferase and aspartate aminotransferase levels [95]. These authors suggest that one possible mechanism is associated with increased transsulfuration of homocysteine, with quercetin acting as a prooxidant [95].
Several randomized control studies were conducted to examine whether homocysteine-lowering B-vitamin therapy would decrease the occurrence of adverse cardiovascular events. Toole, et al. [96] observed that moderate reduction of total homocysteine had no effect on vascular outcomes during the 2 years of follow-up.
Work of Towfighi, et al. [97] observed that age modified the association between B vitamin therapy and recurrent vascular risk in individuals having hyperhomocysteinemia. In this study, older individuals were more likely to benefit from this therapy when compared with younger ones [97].
In the same way, a study of Nigwekar, et al. [98] intended to evaluate the benefits and harms of some of the described homocysteine lowering therapies on all-cause mortality and cardiovascular event rates in patients with end-stage kidney disease. Although these strategies can help in lowering plasma homocysteine levels, these researchers concluded that they did not reduce mortality (cardiovascular and all-cause) or cardiovascular events among these patients.
Several meta-analysis studies have been published evaluating the impact of homocysteine-lowering interventions (vitamins B6, B9 and B12 supplementations) on cardiovascular events and observed no differences in effects of such interventions given alone or in combination on myocardial infarction, death from any cause or adverse events [99-102].
High level of plasma homocysteine is one of the risk factors contributing to cardiovascular disease. Plasma homocysteine content is influenced by diverse genetic and environmental factors. Several hypotheses concerning the pathological mechanism of hyperhomocysteinemia have been proposed, but no clear picture has been established yet.
Hyperhomocysteine influences multiple vascular responses, including coagulation, platelet function, vascular smooth muscle responses, and endothelial function. This condition induces endothelial cell damage and alters the production and/or activity of vasoregulatory mediators like NO. Moreover, high levels of plasma homocysteine impair endothelium-dependent vasodilation and regulation of blood flow, endothelial cell apoptosis that will lead to the development of atherosclerosis and cardiovascular disease.
Treatment strategies aiming the decrease in homocysteine levels have been tested and all point to the correction of coenzymes levels. These coenzymes are essential for maximum activity of enzymes with critical roles in homocysteine metabolism. Supplementations of folic acid and/or vitamins B (mainly B12, but also B6), precursors of the synthesis of the mentioned coenzymes, have been effective in reduction of high levels of homocysteine, in most populations. Another B vitamin, riboflavin, seems to have effect on homocysteine levels in individuals carrying the C677T mutation on the MTHFR gene.
The mechanism of the beneficial effects of folates on the endothelium remains unclear, however it might include antioxidant actions, effects on cofactor availability, or direct interactions with the enzyme endothelial NO synthase [103].