Neurofibromatosis type 1(NF1) is a genetic disorder associated with an increased risk of malignant disorders. Adrenal neuroblastoma is considered an extremely rare tumor in adults and was not previously described in association with NF1.
A 42-year-old normotensive woman with typical signs of NF1 underwent evaluation for abdominal pain, and a large 14 × 10 × 16 cm left adrenal mass displacing the spleen, pancreas and colon was found. An initial diagnosis of pheochromocytoma was done based on the known strong association between pheochromocytoma, NF1 and increased catecholamine levels. The patient underwent surgery in which a left adrenalectomy, nephrectomy and splenectomy were performed.
The pathology examination revealed an adrenal tumor composed of immature neuronal cells intermixed with fibrillar eosinophilic material (neuropil). These features are compatible with a neuroblastoma. No pheochromocytoma component was found in the tumor. Five months after the operation, a vertebral lesion was detected and biopsied with histological findings of metastatic neuroblastoma.
This is a unique case of an adrenal neuroblastoma mimicking a pheochromocytoma in an adult patient with neurofibromatosis type 1.
1. Neurofibromatosis type 1, an autosomal dominant disorder is associated with a known substantial increased risk of developing adrenal pheochromocytomas but not with adrenal neuroblastomas.
2. This is the first reported case of an adrenal neuroblastoma occurring in an adult patient with NF1 presenting as a large adrenal mass with increased catecholamine levels mimicking a pheochromocytoma.
3. This case demonstrates the clinical overlap between pheochromocytoma and neuroblastoma.
Adrenal neuroblastoma, Neurofibromatosis type 1, Pheochromocytoma, Neural crest-derived tumors
Neurofibromatosis type 1 (NF1) is a genetic disorder that involves autosomal dominant mutations in the neurofibromin (NF1) gene [1]. These mutations increase the risk of a variety of tumor types, including benign and malignant tumors [1]. Neuroblastoma (NB) is a malignant neoplasm that is embryologically derived from neural crest cells and therefore develops in any organ of the sympathetic nervous system. NB frequently appears in children, and very rarely in adults. Only 190 adolescent and adult cases of neuroblastoma have been described in the literature from 1987 to 2014 [2], and an abdominal primary tumor site was found in only a quarter of the cases. Adult-onset NB is usually classified as a high-risk tumor and is associated with a poor prognosis [2]. The current study presents a case of a giant catecholamine secreting adrenal neuroblastoma in a patient with NF1. Adrenal neuroblastomas are considered an extremely rare tumor in adults and have not been previously described in association with NF1.
A 42-year-old normotensive woman with neurofibromatosis type 1 (NF1) with typical cutaneous manifestations (multiple neurofibromas, café-au-lait spots and axillary freckling), underwent evaluation for abdominal pain and a large left adrenal mass displacing the spleen pancreas and colon, was found upon a CT scan (Figure 1a). A subsequent fluorodeoxyglucose positron emission tomography-computed tomography (FDG-PET-CT) scan showed a 14 × 10 × 16 cm mass with a high and heterogeneous uptake, areas of necrosis and no evidence of distant metastasis (Figure 1b). Laboratory work-up revealed increased urinary levels of normetanephrine (1160 μg/24 hours, normal < 604 μg/24 hours) and dopamine (1481 μg/24 hours, normal < 400 μg/24 hours). An initial diagnosis of pheochromocytoma was made based on the known strong association between pheochromocytoma, NF1 [1] and a positive biochemical work-up, although the discrepancy between the tumor’s large size and the modest catecholamine elevation was puzzling.
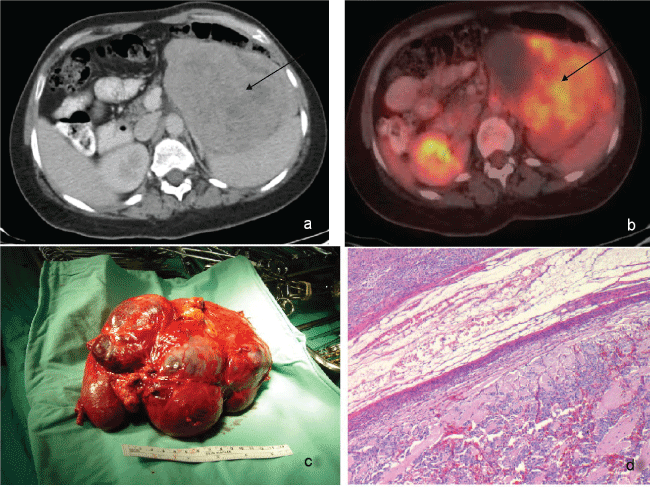 Figure 1: a) Coronal contrast enhanced CT of the abdomen showing a large 14.0 cm adrenal mass displacing the spleen, pancreas and colon (black arrow); b) An FDG PET/CT image shows uptake with high heterogeneous uptake and areas of necrosis (black arrow); c) Surgical specimen; d) Adrenal neuroblastoma (HE stainx40), small round blue immature neuronal cells admixed with eosinophilic fibrillar material (neuropil).
View Figure 1
Figure 1: a) Coronal contrast enhanced CT of the abdomen showing a large 14.0 cm adrenal mass displacing the spleen, pancreas and colon (black arrow); b) An FDG PET/CT image shows uptake with high heterogeneous uptake and areas of necrosis (black arrow); c) Surgical specimen; d) Adrenal neuroblastoma (HE stainx40), small round blue immature neuronal cells admixed with eosinophilic fibrillar material (neuropil).
View Figure 1
Despite normal blood pressure levels in ambulatory 24-hour blood pressure monitoring, the patient was treated preoperatively accordingly to a pheochromocytoma diagnosis with alpha- blockers and subsequently underwent an operation in which a left adrenalectomy, nephrectomy and splenectomy was performed as the tumor involved these organs without a true plane of dissection.
A 16 × 10 × 9 cm adrenal tumor with areas of hemorrhagic necrosis was present (Figure 1c). The spleen and kidney were adherent to, but not involved with the tumor. On microscopic examination, the tumor was composed of a majority of small immature neuronal cells, and a smaller component of neuronal cells in intermediate and late (ganglion cells) differentiation stages. The neuronal cells were embedded in a rich background of fibrillar eosinophilic material (neuropil) (Figure 1d). Despite performing extensive sampling, no pheochromocytoma components were found in the tumor. Upon immune histochemical staining, the tumor cells were positive for chromogranin, synaptophysin, neurofilament, and vimentin. There was focal S100 positivity in sustentacular cells. These findings were compatible with a neuroblastoma.
The patient's recovery after tumor resection was uneventful. There was no evidence of a metastatic disease on the. 123I-Metaiodobenzylguanidine.(123I-MIBG) single photon emission computed tomography (SPECT) imaging two months after the surgery and according to the INSS system, the tumor was defined as stage 1 Three months later, the patient complained of bone pain and a CT scan detected a single D2 vertebral lesion. The biopsy revealed a metastatic neuroblastoma. The patient was treated by local radiotherapy and the symptoms improved. For the next two years, there was no evidence of metastases on serial total body CT scans performed every six months. At that point the bone pain reoccurred and despite normal serial CT scans, an additional MIBG scintigraphy was performed demonstrating multiple bone and liver metastases. Disease stabilization was achieved by chemotherapy treatment with Etoposide and Carboplatinum. Subsequent serial MIBG scintigraphs showed no further disease progression.
Neuroblastoma and pheochromocytoma are both neural crest-derived tumors but current literature describes major differences in their clinical characteristics:
1. Neuroblastomas account for more than 7% of childhood cancers but are considered extremely rare in adults, while pheochromocytomas mostly occur in adulthood [2-4].
2. Only 1-2% of neuroblastoma cases are known to be related to hereditary syndromes, while 15-30% of pheochromocytomas are part of hereditary tumor syndromes [3,5].
3. The most typical presentation of a neuroblastoma is related to its mass effect, while pheochromocytomas are mostly recognized by symptoms and signs caused by excess catecholamines [3]. Despite these differences, recent bioinformatic analyses of genomic data have revealed several similar expression patterns in pheochromocytomas and neuroblastomas [6].
We describe a patient with NF1, an autosomal dominant disorder associated with loss-of-function of the NF1 suppressor gene encoding neurofibromin [1,7]. NF1 patients have a substantially increased risk of developing benign and malignant tumors of neurogenic and non-neurogenic origins [1,7]. Compared with the general population, the prevalence of pheochromocytoma is 1000-to 2000-fold higher in NF1 patients (2% vs. 1-2/100,000) but the age of diagnosis, location and malignancy risk are similar [3,7]. However, according to the existing literature, the association between neuroblastoma and NF1 is rare [7,8], and to the best of our knowledge this is the first reported case of an adrenal neuroblastoma in an adult NF1 patient. Interestingly however, 23% of the rare cases of composite pheochromocytoma that include ganglioneuroblastoma, ganglioneuroma or neuroblastoma, occur in NF1 and a role of neurofibromin in the abnormal proliferation of Schwann cells has been suggested [9].
This is the first report of an adrenal neuroblastoma occurring in an adult patient with NF1 presented as a large adrenal mass with increased catecholamine levels mimicking a pheochromocytoma. This case demonstrates the clinical and possible hereditary overlap between these tumors.
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the reported research.
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Written informed consent was obtained from the patient for publication of the submitted article and accompanying images by her signature on the consent form.
Dr. Harbi Khalayleh researched the data, wrote the manuscript, contributed to the discussion, and reviewed and edited the manuscript. Dr. Hilla Knobler researched the data, wrote the manuscript, contributed to the discussion, reviewed and edited the manuscript. Dr. Vitaly Medvedovsky researched the data, wrote the manuscript and contributed to the discussion. Dr. Edit Feldberg performed the pathology investigation, contributed to the discussion and reviewed and edited the manuscript. Dr. Judith Diment performed the pathology investigation, contributed to the discussion and reviewed and edited the manuscript. Dr. Lena Pinkas reviewed the ¹²³I-MIBG SPECT and FDG PET/CT images Dr. Guennadi Kouniavsky researched the data, wrote the manuscript, contributed to the discussion and reviewed and edited the manuscript. Dr. Taiba Zornitzki researched the data, wrote the manuscript, contributed to the discussion and reviewed and edited the manuscript.