Background: The aim of our study is to evaluate the patients with adrenal incidentaloma in terms of clinical and endocrinological features and, to determine whether a correlation between the malignancy and preoperative tumor size via analyzing of the histopathological results of the masses.
Methods: This study was performed on 320 patients (86 males, 234 females) with adrenal incidentaloma who visited our clinic during a period of 3 years. Chi-square test was used to categorize the patients according to their hormonal status. The relationship between the malignancy and tumor size was examined by correlation analysis method. A p < 0.05 was considered statistically significant.
Results: The mean-size of adrenal mass in men was found higher than women (p < 0.05). The rate of the patients with functional adrenal mass among all cases was 17.5%. Both frequency of pheochromocytoma and malignancy were high in the patients with adrenal mass size > 60 mm (p < 0.05).
Conclusion: We found that the ratio of malignancy was 6.9% in the patient with adrenal mass size ≤ 60 mm. The histopathological results of the patients in our study have suggested that may be approached more conservatively while taking a decision for the surgical excision according to the size of the mass.
Adrenal incidentaloma, Pheochromocytoma, Subclinical Cushing's syndrome, Nonfunctional adrenal mass
CT: Computed Tomography; MRI: Magnetic Resonance Imaging; DST: Dexamethasone Suppression Test; SCS: Subclinical Cushing's Syndrome; ACTH: Adrenocorticotropic Hormone; DHEAS: Dehydroepiandrosterone Sulfate; MN: Metanephrine; NMN: Normetanephrine
Adrenal incidentalomas are incidentally discovered masses detected by imaging modalities performed for purpose other than imaging the adrenal glands, in patients without signs of adrenal diseases. Adrenal incidentalomas have a prevalence of at least 5% in the general population [1]. With the widespread use of imaging techniques, adrenal incidentalomas will be diagnosed more frequently, but also might put burden on the patient due to uncertainty and will have increasing economic consequences for the health system. Therefore, a careful and methodical approach should be attempted in evaluation of these masses. The vast majority of these masses are non-hypersecreting adrenocortical adenomas, although they may represent primary or metastatic malignancies or show various endocrine abnormalities with subclinical hyperfunction.
The differentiation between benign and malignant lesions is the first critical issue and the possibility of malignancy should be considered for all adrenal masses. Although malignancy is often difficult to exclude, it should be primarily suspected with increased age, weight loss, large mass and bilateral masses. In particular, a diameter above 6 cm is significantly associated with malignant tumors [2].
The other major point to address is whether these masses, which are usually asymptomatic, are indeed secretory because some of the adrenal tumors may lead subclinical forms of various hormonal abnormalities including Cushing's syndrome, pheochromocytoma, hyperaldosteronism, or hyperandrogenism. The object of this study is to evaluate the clinical, endocrinological and histopathological characteristics of adrenal incidentalomas, and to propound a strategy for the management of adrenal incidentalomas in the light of our experiences.
This study was performed on retrospectively 320 patients (86 male, 234 female) with adrenal incidentaloma who visited Ataturk Training and Research Hospital, Department of Endocrinology. All the procedures were approved by the institutional research ethics committee before the beginning of the study (Izmir Katip Celebi University Non-invasive Clinical Research Ethics Committee. Prof. Dr. Recep Sutcu, the Chairperson of the Ethics Committee. Protocol number 57, and date of approval October 19, 2012).
The patients who have with a history of malignancy were not included to the study. In addition, the patients with symptoms and/or findings suggestive of any adrenal disease before the radiological examinations were also excluded from the study.
All of the blood analyses were performed at the Laboratory of Hormone and Biochemistry in our hospital. The serum levels of potassium were measured with ion- selective electrode method using the Abbott Architect c16000 instrument. The serum and plasma levels of hormones were determined using a chemiluminescent immunometric assay with an Advia Centaur XP (Siemens, Ireland/U.S.) analyzer. All of the 24-hour urine analyses were performed at the Duzen Laboratories Group, Ankara. The 24-hour urinary fractionated metanephrines and normetanephrines were measured with liquid chromatography-mass spectrometry using WATERS Quattro Premier XE instrument, the levels of free cortisol in 24-hour urine were determined using a electrochemiluminescence immunoassay with a Roche COBAS analyzer. The radiological results of the cases were obtained from measurements using GE Logiq A5 ultrasound, Toshiba Aquilion 64 computed tomography (CT), GE Signa HDxt 1.5T Magnetic Resonance Imaging (MRI) and Philips Intera 1.5T MRI devices at Department of Radiodiagnostic in our hospital. The histopathological examinations of the patients were also performed at Department of Pathology in our hospital. The patients who were evaluated in the study were divided into the groups according to their adrenal mass sizes and hormonal activities. The largest measurement at first detection sizes of the adrenal masses was recorded as the mass size. In the patients with bilateral adrenal masses, the size of the larger mass was taken basis.
Serum cortisol levels were measured both basal and after overnight 1 mg Dexamethasone Suppression Test (DST), and Subclinical Cushing's Syndrome (SCS) was ruled out if serum level of cortisol was less than or equal to 1.8 μg/dL after overnight DST. Two-day, low dose (2 mg) DST was performed in the patients who were could not been excluded SCS by overnight DST. Furthermore, plasma level of adrenocorticotropic hormone (ACTH), level of 24-hour urinary free cortisol, and diurnal changes in serum cortisol concentration (at 11 pm and 7 am) were also measured in these patients. SCS were diagnosed the patients who have at least two of the disorders listed below; non-suppressed serum cortisol level after 2-day 2 mg DST, plasma level of ACTH below 5 pg/mL, impaired diurnal cortisol rhythm, and high level of urinary free cortisol. The 24-hour urinary fractionated metanephrines and normetanephrines were measured in all cases, and the cases which were detected two-fold increase above the upper limit in at least one of the catecholamine metabolites were included to the pheochromocytoma group. In addition to, the patients were assessed in terms of both primary aldosteronism (firstly by measurement of blood pressure and serum potassium concentration, if necessary by measurement of plasma renin activity and aldosterone concurrently) and adrenal hyperandrogenism (by measurement of serum Dehydroepiandrosterone Sulfate [DHEAS] concentration). As a result of all examination, the adrenal masses which were not detected any hormonal hyperactivity were assumed non-functional.
The SPSS 16.0 (SPSS, inc. Chicago, IL) program was used for statistical analysis of the data. Chi-square test was used to categorize the patients according to their hormonal status. The relationship between the malignancy and tumor size was examined by Pearson's correlation analysis method. The data were shown as mean ± standard deviation and values p < 0.05 were accepted statistically significant.
Three hundred and twenty patients were included in the study. The average follow-up period was 26.9 (3-36) months. Two hundred and thirty-four of the cases (73.1%) were women. The mean age of the patients was 55.5 ± 11.8 (16-85) years, there was no significant difference in age between men and women. Majority of cases was in 6th and 7th decades in both genders (Figure 1). The adrenal masses were imaged at right side in 155 cases (48.4%), at left side in 134 cases (41.9%), and bilateral in 31 cases (9.7%).
 Figure 1: The distribution of the cases by age groups.
View Figure 1
Figure 1: The distribution of the cases by age groups.
View Figure 1
The average of mass size of all patients was 30.1 ± 21 (8-170) mm. Mean mass size of men (34 ± 23 mm) was significantly higher than women (28.6 ± 20 mm) (p = 0.002). All cases were divided into three groups in according to age (< 40 years, 40-65 years, > 65 years) as well as tumor size (< 40 mm, 40-60 mm, > 60 mm). The average size of adrenal mass (39.6 mm) in patients under the age of 40 was significantly larger than other (40-65 and > 65 years) patients (29.1 and 29 mm, respectively) (p = 0.003).
The patients were also classified in three groups according to hormonal activity of adrenal masses; the groups of non-functional, SCS, and pheochromocytoma that consisted of from 264 (82.5%), 37 (11.6%), and 19 (5.9%) patients, respectively (Table 1).
Table 1: The distribution of the cases according to hormonal activity and mass size. View Table 1
The vast majority of the size of non-functional masses was less than 40 mm in both genders. The number of case with pheochromocytoma among the patients with adrenal mass larger than 60 mm was significantly higher than other two groups (p = 0.019) (Figure 2). Additionally, the distribution of the cases according to hormonal activity, age, and gender was shown in Table 2.
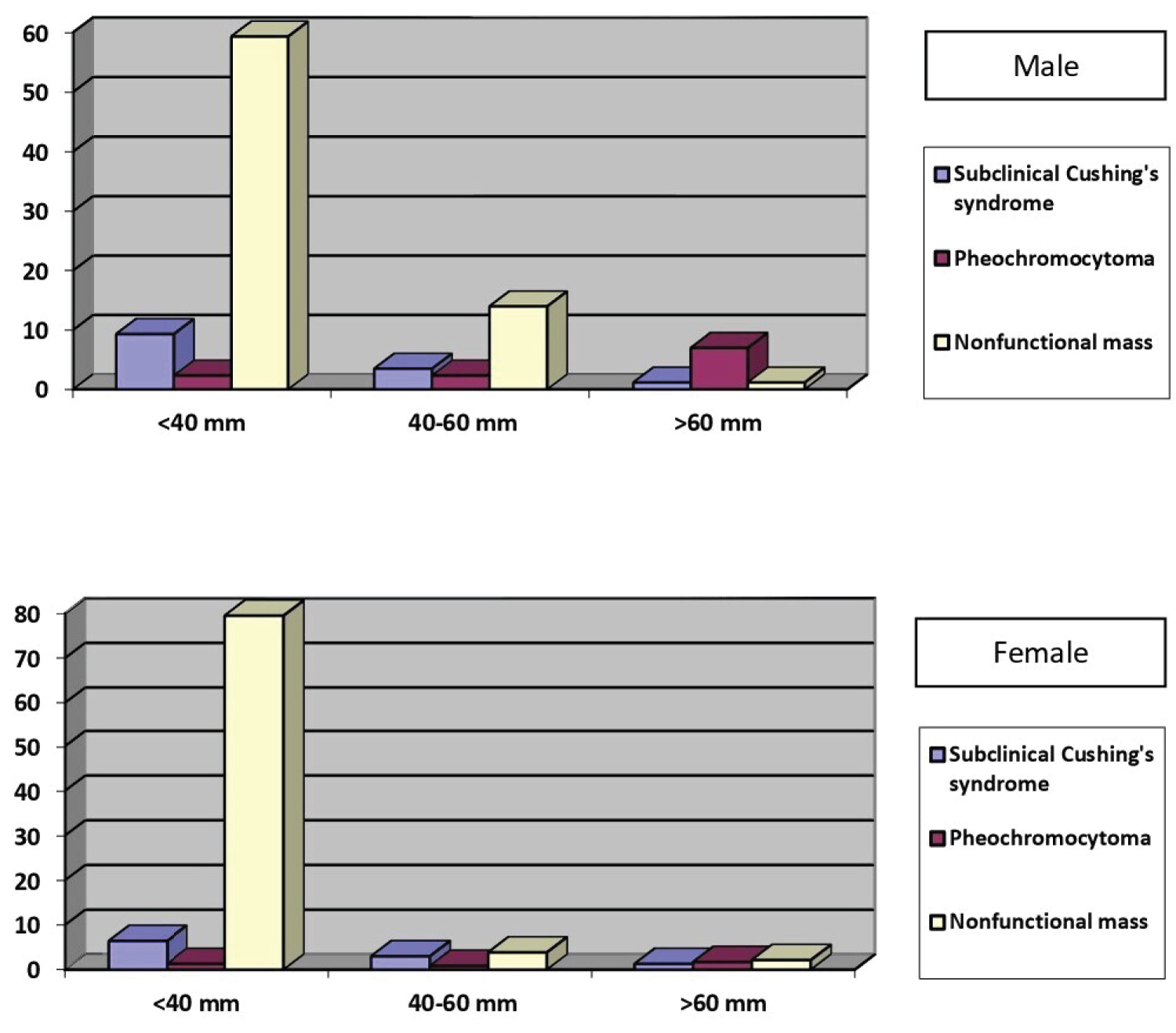 Figure 2: The distribution of the cases according to mass size and hormonal activity.
View Figure 2
Figure 2: The distribution of the cases according to mass size and hormonal activity.
View Figure 2
Table 2: The distribution of the cases according to hormonal activity and age. View Table 2
The non-functional masses that have the sizes of smaller than 40 mm had been followed without surgery, on the other hand the surgery had been recommended the patients with adrenal mass larger than 40 mm even their masses were non-functional. The surgery had been recommended to 23 of 37 cases with SCS, remaining 14 patients had been followed without surgery because none of them had the metabolic complications associated with hypercortisolism such as hyperglycemia, hypertension, obesity, and osteoporosis. All of the cases with pheochromocytoma had been referred to surgery, however, one elderly patient with pheochromocytoma had refused the operation because of the high surgical risk.
The cases who had not undergone surgery were re-evaluated by radiological and functional work-up 6 and 12 months after diagnosis, and then at 1-year intervals. These cases had been followed-up by radiodiagnostic modalities including MRI (72.8%), CT (25.5%), and ultrasound (1.7%). During radiological follow-up, the enlargement at least 10 mm in the sizes of masses had been detected in 20 patients and the surgery had been recommended to 10 of them. It had been decided to continue follow-up in 6 of remaining 10 patients because their adrenal masses were both non-functional and smaller than 40 mm still. The last 4 patients had been diagnosed extra-adrenal malignancy in the course of the investigations and their adrenal masses had been considered as metastasis of primary malignancies. None of the patients had been shown alteration of their hormonal status throughout follow-up.
The histopathological examination had been performed in 52 patients, the tissue materials had been obtained by tru-cut biopsy in 2 cases, intraoperative biopsy in 1 case, and cytological sampling in 1 case. Forty-eight patients had surgery, the operations had been carried out successfully in 47 of them but in the remaining one case, the adrenal mass which was adherent to the aorta could not be excised completely. Histopathological examinations revealed benign lesions in 40 cases (76.9%) and malignant tumors in 12 cases (23.1%) (Figure 3).
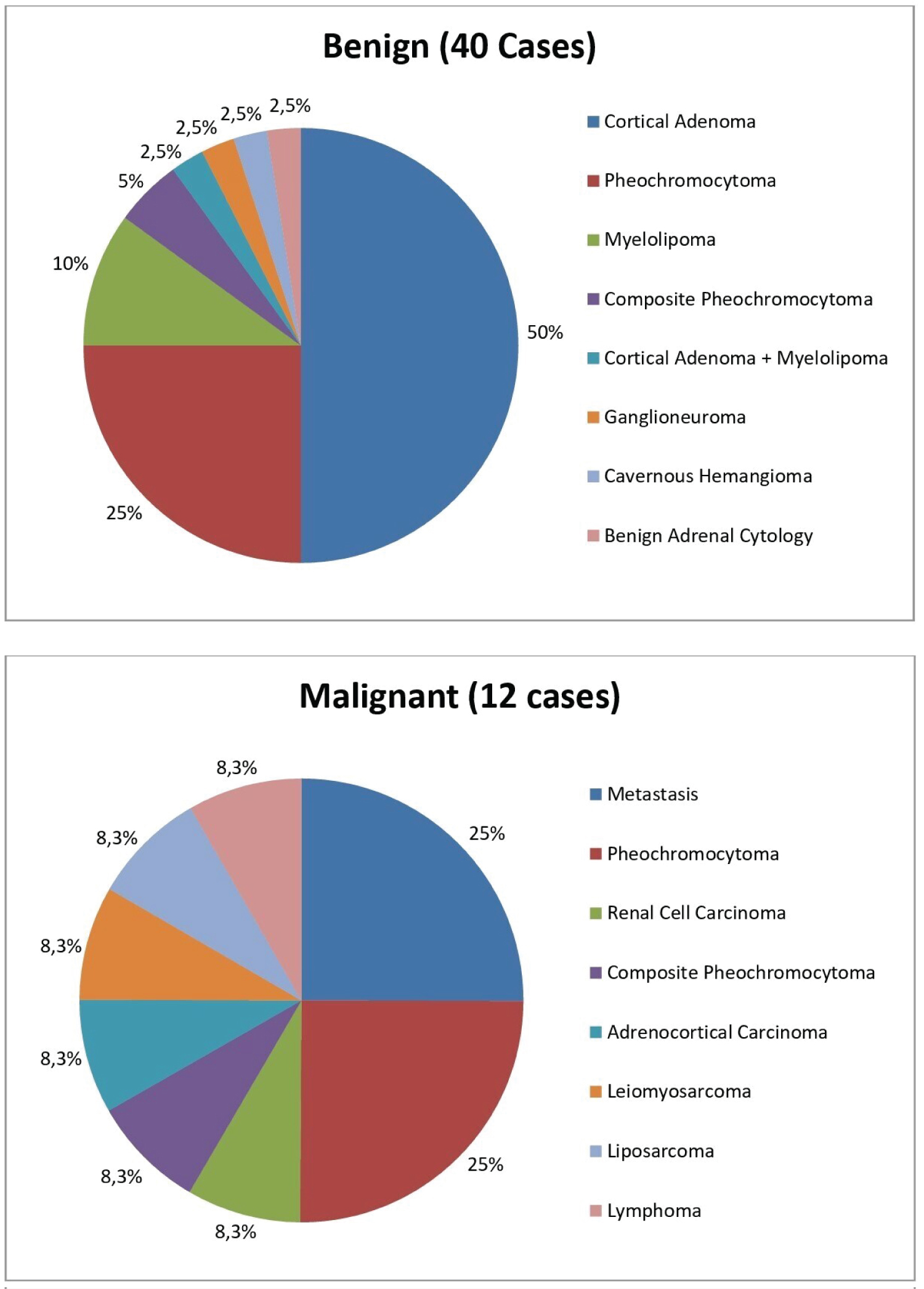 Figure 3: The distribution of the cases according to histopathological results.
View Figure 3
Figure 3: The distribution of the cases according to histopathological results.
View Figure 3
According to the results, there was significant correlation neither between age and malignancy (p = 0.233) nor between gender and malignancy (p = 0.841). On the other hand, there was a significant correlation between mass size and malignancy. The rate of malignancy was found 43.5% among the patients with adrenal mass larger than 60 mm, whereas malignancy was detected in only 2 of 29 (6.9%) cases with adrenal mass size of smaller than 60 mm (Table 3).
Table 3: The distribution of the histopathological results according to mass size. View Table 3
The widespread use of imaging procedures has led to an increased discovery of incidental masses in the adrenal glands. Adrenal incidentalomas have a female sex predilection. Likewise, our study has also shown that these masses had female preponderance. On the other hand, there is no evidence of a sex difference in prevalence from autopsy studies or general health exams [3]. This result probably reflects the sex distribution of the population undergoing imaging procedures and it might be explained by a referral bias (more imaging studies are recommended for women due to various reasons). However, adrenal incidentalomas seem to be related to age. The frequency of incidentaloma increases from about 0.2 to 7% from youth to old age [4]. It has been suggested two factors that cause increasing with age in the incidence of adrenal incidentaloma: One of them is nodular regenerative response of the adrenal tissue which expose to chronic ischemia, the other is more imaging studies in the elderly due to various comorbidities. The patients in 6th and 7th decades had constituted the vast majority of cases in our study consistent with the literature [5].
It is known that there is a positive correlation between the adrenal mass size and malignancy; hence some authors recommend excision of lesions greater than 40 mm [6,7]. Concerning the size of the adrenal masses, it is impossible to find a threshold warranting that a lesion would be benign. The stability of a lesion signifies benignity because it is highly unusual for untreated malignant lesions to remain stable for more than 6 months [8]. Furthermore, while evaluating the malignancy possibility of an adrenal mass by imaging techniques should be taken into account its size as well as homogeneity, presence of necrosis and calcifications and extent of invasion. However, if CT characteristics point to a myelolipoma or cyst, and the patient is asymptomatic, nonsurgical management is appropriate, regardless of mass size [9]. Lipid-sensitive imaging techniques are extremely useful for the characterization of adrenal incidentalomas since up to 70% of adrenal adenomas contain abundant intracellular fat, whereas almost all malignant lesions are lipid poor [10]. Chemical shift MRI, similar to CT densitometry, relies on the detection of intracellular lipid in an adenoma for accurate adenoma detection [11]. Generally, adenomas enhance rapidly following intravenous contrast administration and have prompt contrast excretion - a phenomenon termed ‘contrast washout'. Malignant lesions also enhance rapidly but usually have slower washout due to capillary leakage. A 15-min delayed CT protocol, applying an absolute percentage washout of 60% or more, has a sensitivity of 86-88% and a specificity of 92-96% for the characterization an adrenal adenoma [12,13]. Despite all these, unfortunately, benign and malignant masses cannot be precisely discriminated on the basis of radiological features alone. In this study, surgical excision had been recommended for non-functional adrenal masses greater than 40 mm in addition to hyperfunctional masses regardless of their sizes. In histopathological results of the operated cases, malignancy rate was detected about 7% for adrenal masses with sizes smaller than or equal to 60 mm, moreover none of the adrenal masses with sizes between 40 and 60 mm was malignant. Therefore, it may be a conservative change in our clinical approach after this.
Clinically inapparent adrenal masses are most often non-functioning tumors. Several studies have elucidated the characteristics of adrenal incidentalomas, showing that 58%-86.2% of adrenal masses were non-functioning, 6%-10.9% were associated with SCS, 2.1%-20% were pheochromocytomas, and 1.6%-10% were primary aldosteronism [13-16]. In our study, the percentages of non-functional masses, SCS, and pheochromocytoma were detected about 82.5%, 11.5%, and 6% respectively. It may be uncommon that there was no case with primary aldosteronism in a series of 320 patients with adrenal incidentaloma. In fact, there was a female patient with hypertension and persistent hypokalemia in our study. Unfortunately, primary aldosteronism could not be demonstrated in this patient because plasma renin activity and serum aldosterone concentration could not be measured for technical reasons. Thus, the patient was not be included the study although she had clinical suspicion of Conn's syndrome. Thereupon, the cases with primary aldosteronism that are followed-up in our clinic were reviewed and it was noticed that their adrenal masses had been detected during investigation of the etiology of hypokalemia in all cases. Therefore, these cases were also excluded from our study. Why there was no case with primary aldosteronism in our study may be explained that most of the patients with primary aldosteronism are actually diagnosed in course of tests for hypokalemia.
Sex hormones secreting adrenocortical tumors are rare and typically have clinical features of androgen or estrogen excess [17]. High level of DHEAS may be an evidence of adrenal androgen excess. Additionally, the measurement of DHEAS may also contribute to the differential diagnosis of malignant masses. Because serum DHEAS levels are frequently elevated in patients with clinically manifest adrenocortical carcinoma. As a matter of fact, except for adrenocortical carcinoma, endocrinological evaluation does not contribute very much to the diagnosis of malignant tumors. Lee, et al. have been suggested that since all virilizing and feminizing tumors are symptomatic, screening for androgen or estrogen excess in patients with an adrenal incidentaloma is, in fact, unnecessary [18]. The results of our study have also supported this opinion. The concentration of serum DHEAS had been detected 5491 mcg/dL (normal ranges for age; 35-430) in a female patient in SCS group of our study. The symptoms associated with hyperandrogenism of the patient had dramatically decreased after the excision of her adrenal mass. Thereupon, we have considered that her adrenal mass had secreted not only cortisol but also DHEAS. Moreover, diameter of the mass was 10 cm but its histopathological result was benign (adrenocortical adenoma) contrary to expectations.
Adrenocortical carcinoma is an uncommon neoplasm with an estimated annual incidence of 0.5-2 cases per million population [19]. Nevertheless, it is essential to diagnose early as possible as because the tumor has very aggressive course. The prevalence of primary adrenocortical carcinoma is clearly related to the size of the tumor. The risk increases with enlarging the diameter of lesion even though malignant tumors smaller than 3 cm in diameter have occasionally been described [2,13]. Unfortunately, there are no reliable markers to distinguish adrenocortical carcinoma from adenoma. Even fine needle aspiration biopsy taken from an adrenal mass cannot provide the differential diagnosis between carcinoma and adenoma. Moreover, biopsy of an adrenocortical carcinoma may result seeding of tumor cells along the needle track [20]. There was only one patient with adrenocortical carcinoma in our study. The patient had both laboratory findings of pheochromocytoma according to base on the reference range in this study and an adrenal mass with diameter of 9 cm, she has been followed postoperatively without recurrence or metastasis since 18 months.
Pheochromocytoma is a rare tumor arising from chromaffin cells of the adrenal medulla or in the paraganglia that produce excessive amounts of catecholamines [21]. Because of catecholamines are metabolized within chromaffin cells to metanephrine (MN) or normetanephrine (NMN), these metabolites can be used to diagnose pheochromocytoma [22]. Furthermore, assays of MN and NMN in plasma or urine have better diagnostic sensitivity than measurements of the parent catecholamines; therefore, determination of plasma or urine MN and NMN is recommended over other biochemical indices [23]. In our study, the measurements of MN and NMN in 24-hour urine had been used to diagnose pheochromocytoma. Eighteen patients had been operated for suspicion of pheochromocytoma. The diagnosis of pheochromocytoma had been confirmed in 15 of these patients after surgical excision. Adrenocortical adenoma, adrenocortical carcinoma, and metastasis of renal cell carcinoma had been detected in the remaining 3 patients, respectively. The histopathological result of a patient who had normal levels of 24-hour urine MN and NMN was composite pheochromocytoma (pheochromocytoma + myelolipoma). According to these results; sensitivity, specificity, positive predictive value, negative predictive value, and validity of the method which were used to diagnose pheochromocytoma have been calculated as 93.75%, 91.67%, 83.3%, 97.06%, and 92.3%, respectively. The relative decrease in specificity of the method may due to that the threshold for the laboratory diagnosis of pheochromocytoma was adopted over two-fold of the upper limit. Thus, Eisenhofer, et al. have suggested that it should be 4-fold or more increase in the levels of catecholamine metabolites to suspect pheochromocytoma [24].
The effective treatment of pheochromocytoma is completely removal of the mass by surgery. However, the treatment of the patients with SCS is still controversial. Data from high-quality prospective trials are lacking to guide the optimal management of SCS and to indicate the superiority of a surgical or a non-surgical approach [25,26]. Until the risks and benefits of adrenalectomy are elucidated, it seems reasonable to elect for surgery younger patients with SCS who display some of diseases (hypertension, diabetes, obesity, and osteoporosis) potentially attributable to excessive cortisol exposure that are of recent onset, or are resistant to optimal medical treatment, or are rapidly worsening [27,28]. Although the natural history of SCS and its morbidity are unclear, we also advocate the reasonability of adrenalectomy for patients with this condition, especially in the presence of metabolic disturbances (hypertension, obesity, diabetes or osteoporosis) potentially aggravated by glucocorticoid excess. Therefore, the surgery had been recommended to most of our cases with SCS for their adrenal masses. Since the hypothalamo-pituitary-adrenal axis and the contralateral adrenal gland may be suppressed by prolonged cortisol secretion, the patients with SCS also require glucocorticoid therapy both during and following surgery similarly the patients with overt Cushing's syndrome. Because of increased cardio-metabolic risk in SCS patients, the cases that are not operated on should be followed under the aggressive medical treatment for metabolic components of the syndrome.
It has been reported that the hormonal functions of adrenal incidentalomas might change in course of time [29]. However, our data have demonstrated that none of patients showed alteration in point of their hormonal status throughout follow-up. Most endocrine authorities recommend annual biochemical testing for 2-4 years. However, the risk of an adrenal incidentaloma, initially thought to be non-functional, subsequently developing clinically important endocrine functionality over 2-4 years is less than 1%. Kastelan, et al. have also reported that the risk of an adrenal mass initially diagnosed as benign and non-functional becoming malignant or hormonally active is extremely low [30]. In a recent study, it has been suggested that patients with an initial normal hormonal screening may not need further biochemical follow-up [31]. Additionally, Schalin-Jäntti, et al. have suggested that small lipid-rich adrenal incidentalomas did not demonstrate excessive growth or hormonal hypersecretion on 5-year follow-up and it may be sufficient to perform control CT and screen for hypercortisolism after 5 years in such incidentalomas [32]. On the other hand, Bulow, et al. have reported a female patient with non-functioning adrenal incidentaloma at discovery but she has been diagnosed pheochromocytoma both hormonally and histologically after 9 years of observation [33].
During the follow-up period, twenty patients had showed an increase in tumor size of more than or equal to 1 cm in our series, and 10 of these patients had undergone surgery. Malignancy was proved histologically in two patients (one of them was leiomyosarcoma, the other was lymphoma), 4 patients without surgery were diagnosed extra-adrenal malignancy in the course of the investigations and their adrenal masses were considered as metastasis of primary malignancies. All of the mass enlargements had been detected in the first year of follow-up period, so we have suggested that imaging follow-up should be repeated in 6 and 12 months after diagnosis. Nevertheless, a limitation of our study is that approximately one-third of the cases had less than 1 year of both imaging follow-up and endocrinological follow-up, thus the nature of their adrenal lesions is unknown.
The most commonly used imaging modality in assessing adrenal incidentaloma is CT. X-ray imaging techniques, including CT, involve ionising radiation, which carries an associated risk of inducing malignancy. There is direct epidemiological evidence from human populations demonstrating that acute exposure to ionising radiation at doses in the 10-50 mSv range (i.e. the organ dose range typically delivered by two or three CT scans) increases the risk of some cancers [34]. Therefore, in our series, MRI had been preferred as the imaging modality during the follow-up of more than 70% of the patients.
Consequently, surgical excision is essential for large and suspicious masses whether or not hypersecretory states. On the other hand, the follow-up carefully may be eligible for masses of smaller size and those which are hormonally inactive to detect any potential change of status in the future period.
In conclusion, our data indicate that a conservative management is reasonable in the majority of incidentalomas. Indeed, the risk of malignancy seems low (6.9%) for adrenal incidentalomas in size of less than 60 mm even in case of a slight increase in mass size.
This study was not supported by any financial sources. There is not any individual who contributed to the study except mentioned authors. No potential conflict of interest relevant to this article was reported.
The authors declare that each author have equal contribution to the manuscript.