Due to the involvement of the imbalance of Ca2+ homeostasis in neuronal cells in the pathogenesis of several neurodegenerative disorders such as Alzheimer's Disease (AD), the use of drugs to prevent or attenuate this imbalance emerged as a new therapeutic perspective for this disease. Then, our recent discovery of the involvement of the interaction between intracellular signaling pathways mediated by Ca2+ and cAMP (Ca2+/cAMP signaling interaction) in the neurotransmission and neuroprotection, and also its pharmacological modulation, opens a large avenue for the drug development for cognitive deficit treatment in AD. This discovery emerged from several clinical studies performed since 1975 that reported that the use of L-type Ca2+ Channel Blockers (CCB) in the antihypertensive therapy decreased arterial pressure, but promoted typical symptoms of sympathetic hyperactivity such as tachycardia and increment of catecholamine plasma levels. Despite these adverse CCB-effects have been initially attributed to adjust reflex of arterial pressure, during almost four decades this enigmatic phenomenon remained unclear. We discovered in 2013 that these adverse effects resulted from increase of transmitter exocytosis from sympathetic neurons and adrenal chromaffin cells stimulated by CCB due to its modulatory action on the neural Ca2+/cAMP signaling interaction. In addition, we discovered that the pharmacological modulation of this signaling can attenuate neuronal death resulting from cytosolic Ca2+ overload due probably to activation of cellular survival pathways mediated by Protein Kinase cAMP-dependent (PKA) and cAMP-Response Element Binding protein (CREB). Then, our discovery of the role of Ca2+/cAMP signaling interaction in neurotransmission and neuroprotection could importantly contribute to the AD therapy.
Alzheimer's disease, Ca2+/cAMP signaling interaction
Clinical studies performed since 1970's have reported that acute and chronic administration of L-type Ca2+ Channel Blockers (CCB) during antihypertensive therapy, such as nifedipine and verapamil, reduces efficiently arterial pressure but produce typical symptoms of the sympathetic hyperactivity such as tachycardia and enhance of catecholamine plasma levels [1]. Despite these adverse CCB-effects initially attributed to adjust reflex of arterial pressure represented a potential risk for the antihypertensive therapy, the mechanisms involved in these enigmatic effects remained unclear for decades.
To investigate the molecular mechanisms involved in the sympathetic hyperactivity induced by CCB, some in vitro studies using isolated smooth muscles richly innervated by sympathetic nerves (rodent vas deferens) to exclude the influence of adjusting reflex showed that the contractile responses of these muscles induced by neurotransmitter release from sympathetic nerves (neurogenic contractions) were significantly reduced or completely abolished by L-type CCB in high concentrations, but unexpectedly and paradoxically potentiated in low concentrations [2-4]. These studies concluded that this in vitro sympathetic hyperactivity induced by CCB (in low concentration) was resultant from the direct action of these drugs on the contractile mechanisms, excluding partially the influence of adjusting reflex stimulated by CCB. However, the precise molecular mechanisms involved in this paradoxical sympathetic hyperactivity induced by CCB, defined by us as "calcium paradox" [5], remained unclear during almost four decades.
Using classical models for in vitro study of the mechanisms involved in the regulation of the sympathetic activity, we discovered in 2013 that the sympathetic hyperactivity induced by CCB was resultant from its modulatory action on the secretory activity of the sympathetic neurons and adrenal chromaffin cells [5]. In the studies performed in an isolated smooth muscle richly innervated by sympathetic nerves (rat vas deferens), we showed that the contractile responses mediated by neurotransmitter released from sympathetic nerves by means Electrical Field Stimulation (EFS) were significantly reduced by L-type CCB (verapamil) in high concentrations (> 1 μmol/L), but paradoxically increased in concentrations below 1 μmol/L, characterizing CCB-induced sympathetic hyperactivity (Figure 1A). This paradoxical CCB-effect was significantly potentiated by pre-treatment of the preparation with the drugs that produced increment of cytosolic cAMP concentration ([cAMP]c), namely as cAMP-enhancer compounds, such as Adenylyl Cyclase (AC) activators (forskolin) and Phosphodiesterase (PDE) inhibitors (rolipram and Isobutyl Methyl Xanthine (IBMX)) (Figure 1B, Figure 1C and Figure 1D). This potentiation was prevented by AC inhibition with SQ 22536, suggesting the involvement of the functional interaction between the intracellular signaling pathways mediated by Ca2+ and cAMP (Ca2+/cAMP signaling interaction) in this response [5].
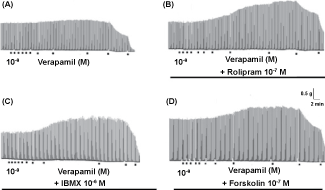 Figure 1: Increase of neurotransmission produced by pharmacological modulation of neural Ca2+/cAMP signaling interaction A) Records showing that contractile responses mediated by neurotransmitter released from sympathetic nerves by means Electrical Field Stimulation (EFS) in rat vas deferens (neurogenic contractions) were significantly reduced by L-type CCB (verapamil) in high concentrations (> 10-6 M), but paradoxically increased in concentrations below 10-6 M, characterizing CCB-induced sympathetic hyperactivity. This increase of neurogenic contractions by verapamil (< 10-6 M) was potentiated by pre-treatment of isolated tissue with cAMP-enhancer compounds, such as rolipram 10-7 M; B) IBMX 10-6 M; C) Forskolin 10-7 M; D) Each point below the record represents molar concentration of verapamil (interval of 0.5 log unity). Each line below the record represents incubation time with cAMP-enhancer compounds. Representative records extracted from [5]. View Figure 1
Figure 1: Increase of neurotransmission produced by pharmacological modulation of neural Ca2+/cAMP signaling interaction A) Records showing that contractile responses mediated by neurotransmitter released from sympathetic nerves by means Electrical Field Stimulation (EFS) in rat vas deferens (neurogenic contractions) were significantly reduced by L-type CCB (verapamil) in high concentrations (> 10-6 M), but paradoxically increased in concentrations below 10-6 M, characterizing CCB-induced sympathetic hyperactivity. This increase of neurogenic contractions by verapamil (< 10-6 M) was potentiated by pre-treatment of isolated tissue with cAMP-enhancer compounds, such as rolipram 10-7 M; B) IBMX 10-6 M; C) Forskolin 10-7 M; D) Each point below the record represents molar concentration of verapamil (interval of 0.5 log unity). Each line below the record represents incubation time with cAMP-enhancer compounds. Representative records extracted from [5]. View Figure 1
As the activity of AC is finely regulated by Ca2+ [6], the reduction of [Ca2+]c produced by L-type CCB results in increase of activity of AC and consequently the elevation of [cAMP]c that which stimulate the cAMP-dependent Protein Kinase (PKA), that in turn activate Endoplasmic Reticulum (ER) Ca2+ channels, such as ER- Ca2+ channels regulated by Ryanodine Receptors (RyR), stimulating Ca2+ release and recruiting of secretory vesicles docked in plasma membrane, and consequently increasing neurotransmitter release and synaptic concentration of neurotransmitters [7-15]. Then, we clearly demonstrated that the reduction of Ca2+ influx through L-type Voltage-Activated Ca2+ Channels (VACC) produced by CCB increases neurotransmitter release and synaptic transmission due to its modulatory action on the Ca2+/cAMP signaling interaction [7-14,16]. This discovery that solved the enigma of almost four decades involved in the sympathetic hyperactivity caused by CCB was published in Cell Calcium in 2013 [5], making it the most accessed article (TOP 1 - full year 2013) of the "Elsevier/ScienceDirect - TOP 25 Hottest Articles (see http://top25.sciencedirect.com/subject/biochemistry-genetics-and-molecular-biology/3/journal/cell-calcium/01434160/archive/50/).
Our studies clearly demonstrated that the pharmacological modulation of neural Ca2+/cAMP signaling interaction by combined use of the L-type CCB (in low concentration) and cAMP-enhancer compounds produces the increase of synaptic transmission due mainly to facilitation of neurotransmitter release mediated by increment of the response of neuronal secretory machinery [7-14]. Our discovery of the role of the Ca2+/cAMP signaling interaction in the neurotransmission and its pharmacological modulation could contribute to development of the new therapeutic strategy to increase neurotransmission in neurodegenerative diseases related to severe deficit in central neurotransmission, such as AD [7-14].
Our discovery of the pharmacological modulation of Ca2+/cAMP signaling interaction allowed explore the participation of this signaling in other cellular responses. It is well established that the cAMP stimulates the cellular survival pathways mediated by PKA and Camp-Response Element Binding protein (CREB) [17-20]. Thus, we proposed that the neuronal death resulting from cytosolic Ca2+ overload associated to neurodegenerative disorders could be attenuated or prevented in response to stimulation of the cellular survival pathways mediated by cAMP/PKA/CREB signaling by means the pharmacological modulation of the Ca2+/cAMP signaling interaction [7-14]. Thus, our discovery of the role of the Ca2+/cAMP signaling interaction in neurotransmission and neuroprotection, and its pharmacological modulation, may open a large avenue for the development of a new therapeutic strategy for neurodegenerative disorders such as AD [7-14,21]. Then, in this review we will discuss how the pharmacological modulation of the Ca2+/cAMP signaling interaction could be a new therapeutic strategy to treat the cognitive deficit in AD.
It is important to note that the growing increase in the life expectancy of the world's population has increased the concern about neurodegenerative disorders related to aging, such as AD. According to a 2015 United Nations report on world population ageing, the number of people aged 60 and older worldwide is projected to more than double in next 35 years, reaching almost 2.1 billion people. Most of this growth will come from developing regions of the world, although the oldest old, who are more than 80 years of age, are the fastest growing segment of the population in developed regions. Despite these improvements in life expectancy, AD have arguably become the most dreaded maladies of older people.
AD is a progressive neurodegenerative disorder related to ageing characterized by cognitive and memory deterioration. Neuritic plaques represent the pathological status of AD, and are respectively related to the accumulation of the β-Amyloid peptide (Aβ) in brain tissues [22,23]. According to the amyloid hypothesis, the overproduction of Aβ is a consequence of the disruption of homeostatic processes that regulate the proteolytic cleavage of the Amyloid Precursor Protein (APP). Genetic and age-related factors could contribute to a metabolic change, favoring the amyloidogenic processing of APP in detriment of the physiological secretory pathway [22,23].
The neurotoxic potential of the Aβ results from its biochemical properties that favor aggregation. These processes, along with a reduction of Aβ clearance from the brain, leads to the extracellular accumulation of Aβ, and the subsequent activation of neurotoxic cascades that ultimately lead to cytoskeletal changes, neuronal dysfunction and cellular death [22]. Intracerebral amyloidosis development in AD patients is in an age-dependent manner, but recent evidences indicate that it may be observed in some subjects as early as in the third or fourth decades of life, with increasing magnitude in late middle age, and highest estimates in old age [22-24].
Therapies targeting the modification of amyloid-related cascades may be viewed as promising strategies to attenuate or even to prevent dementia [22]. Therefore, the cumulative knowledge on the pathogenesis of AD derived from basic science models will hopefully be translated into clinical practice in the forthcoming years. Other targets relevant to AD have also been considered in the last years for producing multitarget compounds [25,26].
In addition to what has been discussed above, Acetylcholinesterase (AChE) is another important target to treat the pathogenesis of AD (cholinergic dysfunction hypothesis). Considering the current hypothesis of accumulation of the Aβ in AD, this relies in the reduction of neurotransmitter Acetylcholine (ACh) release in central cholinergic nervous system involved in cognitive function. Thus, the inhibition of ACh degradation by AChE is a potential target to treat AD [25-27].
An imbalance of intracellular Ca2+ homeostasis also contributes to the pathogenesis of AD [28]. Several evidences suggest that aging impairs ability of the brain intracellular Ca2+ degradation which is likely to induce cellular damage due to cytosolic Ca2+ overload leading to neural death and resultant cognitive dysfunction, such as AD [28]. Therefore, regulation of intracellular Ca2+ homeostasis may represent a new strategy for treatment of AD.
A 10-year follow-up study (2000 to 2010) involving 82,107 hypertensive patients of more than 60 years of age, showed that use of L-type CCB reduced blood pressure and risk of dementia in hypertensives, suggesting that these drugs could be clinically used to treat AD [29]. Supportive findings for the neuroprotective effects of CCB have been showed in 1,241 elderly hypertensive patients with memory impairment [30]. The use of CCB decreased the risk of cognitive impairment and AD independently of blood pressure levels when compared to patients not treated with CCB [30]. The long-term effects of antihypertensive therapy initiated with a long-acting dihydropiridine, such as nitrendipine, has been showed in the double-blind, placebo-controlled Syst-Eur trail in which the incidence of dementia was reduced by 55% [31].
Some studies have proposed that hybrid compounds having the moieties of tacrine, a potent inhibitor of AChE, and nimodipine, a L-type CCB could be useful to the AD pharmacotherapy [25,26]. In addition, galantamine (a moderate AChE inhibitor and a potent allosteric ligand of nicotinic colinoceptors) has been used to improve cognition and behavior in patients with AD [27]. Studies using AD model rats showed that cAMP-enhancer compounds, such as nobiletin (a polymethoxylated flavone from citrus peels) and oxyntomodulin (a proglucagon-derived peptide that co-activates the GLP-1 receptor and the glucagon receptor), produce neuroprotective effects mediated by intracellular cAMP synthesis, activation of PKA and MAPK pathways and phosphorylation of CREB [18,20].
Our discovery of the involvement of the Ca2+/cAMP signaling interaction in the neurotransmission and neuroprotection has produced important advances in the understanding of the pathophysiology and pharmacology of AD [7-14]. These advances allowed us to propose that pharmacological modulation of the Ca2+/cAMP signaling interaction produced by combined use of the L-type CCB (used in the antihypertensive therapy), such as isradipine, and cAMP-enhancer compounds (used in the anti-depressive therapy), such as rolipram, could represent a new therapeutic strategy for treatment of AD in humans. This pharmacological modulation could attenuate cognitive deficit due to increase in central cholinergic neurotransmission caused by increment in ACh release from cholinergic neurons [7-14]. In addition, the pharmacological modulation of the Ca2+/cAMP signaling interaction could reduce or prevent neuronal death caused by cytosolic Ca2+ overload due to increase of [cAMP]c and stimulation of cellular survival pathways mediated by cAMP/PKA/CREB signaling pathway [17-20]. Thus, we have proposed that the pharmacological modulation of this signaling could be a new therapeutic strategy to slow the progression and reduce the symptoms of AD [7-14]. Figure 2 shows how pharmacological modulation of the Ca2+/cAMP signaling interaction could produce increase of neurotransmission and neuroprotection in AD.
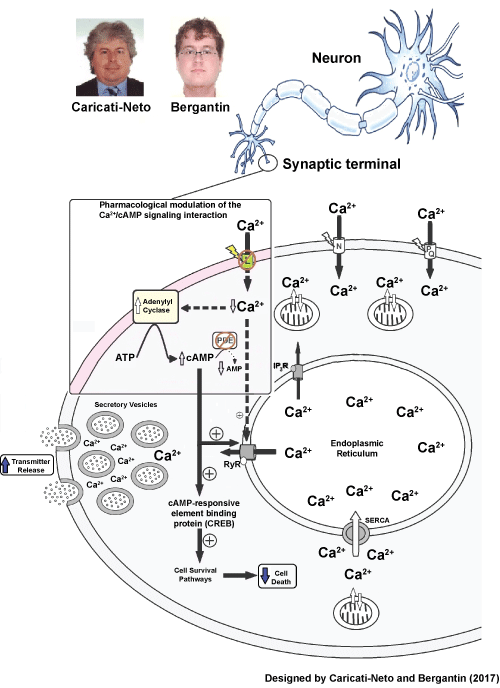 Figure 2: Increase of neurotransmitter release and attenuation of neuronal death (neuroprotection) produced by pharmacological modulation of the Ca2+/cAMP signaling interaction by combined use of L-type Ca2+ Channel Blockers (CCB) and cAMP-enhancer compounds. Figure extracted from [8-14] Schematic representation of the model: Ca2+ entry through L-type voltage-activated Ca2+ channels inhibits Ca2+- sensitive Adenylyl cyclase, inhibiting cAMP signalling pathway- Ca2+ release from the endoplasmic reticulum. Typically, by reducing Ca2+ influx, and consequently [Ca2+]c, L-type CCBs may reduce transmitter release. However, CCBs may also activate Ca2+- sensitive Adenylyl cyclase, activating cAMP signalling pathway-Ca2+ release from the endoplasmic reticulum. Considering there have been described 9 isoforms of Adenylyl cyclase, the isoforms 5 and 6 can be inhibited by Ca2+ [6]. Ca2+ Channel Blockers (CCBs); Ryanodine Receptors (RyR); Mitochondria (MIT); Phosphodiesterase (PDE); Adenosine Tri Phosphate (ATP); cyclic Adenosine Mono Phosphate (cAMP); Adenosine Mono Phosphate (AMP); Endoplasmic reticulum Ca2+-ATPase (SERCA). View Figure 2
Figure 2: Increase of neurotransmitter release and attenuation of neuronal death (neuroprotection) produced by pharmacological modulation of the Ca2+/cAMP signaling interaction by combined use of L-type Ca2+ Channel Blockers (CCB) and cAMP-enhancer compounds. Figure extracted from [8-14] Schematic representation of the model: Ca2+ entry through L-type voltage-activated Ca2+ channels inhibits Ca2+- sensitive Adenylyl cyclase, inhibiting cAMP signalling pathway- Ca2+ release from the endoplasmic reticulum. Typically, by reducing Ca2+ influx, and consequently [Ca2+]c, L-type CCBs may reduce transmitter release. However, CCBs may also activate Ca2+- sensitive Adenylyl cyclase, activating cAMP signalling pathway-Ca2+ release from the endoplasmic reticulum. Considering there have been described 9 isoforms of Adenylyl cyclase, the isoforms 5 and 6 can be inhibited by Ca2+ [6]. Ca2+ Channel Blockers (CCBs); Ryanodine Receptors (RyR); Mitochondria (MIT); Phosphodiesterase (PDE); Adenosine Tri Phosphate (ATP); cyclic Adenosine Mono Phosphate (cAMP); Adenosine Mono Phosphate (AMP); Endoplasmic reticulum Ca2+-ATPase (SERCA). View Figure 2
Our recent discovery of the role of Ca2+/cAMP signaling interaction in neurotransmission and neuroprotection could promote important advances in the pathophysiology and pharmacology of the neurodegenerative disorders. These advances can contribute to drug development more effective and safer to attenuate or prevent cognitive deficit and other clinical symptoms of the AD.
Caricati-Neto and Bergantin thank the continued financial support from CAPES, CNPq and FAPESP (Bergantin's Postdoctoral Fellowship FAPESP #2014/10274-3). The authors also thank Elsevier - "author use": Reuse of portions or extracts from the article in other works - https://www.elsevier.com/__data/assets/pdf_file/0007/55654/AuthorUserRights.pdf.